




Abstract
High‐dose intravenous immunoglobulin (IVIg) is an effective treatment for many antibody‐mediated neuromuscular diseases, suggesting that IVIg directly interferes with the pathways through which the pathogenic antibodies exert their effects. However, the precise mechanisms of action are unclear. Serum anti‐GQ1b antibodies are strongly associated with ophthalmoplegia in patients with Miller Fisher syndrome (MFS) and Guillain–Barré syndrome (GBS). They induce complement‐mediated α‐latrotoxin‐like effects on mouse neuromuscular junctions (NMJs) ex vivo, comprising transient muscle fibre twitching, due to a dramatic increase in the frequency of miniature end‐plate potentials (spontaneous quantal acetylcholine release), followed by transmission block. To clarify the mechanisms by which IVIg may act in MFS and GBS, we investigated its effects on the interaction of anti‐GQ1b antibodies with GQ1b in vitro and on anti‐GQ1b antibody‐mediated NMJ injury ex vivo, using anti‐GQ1b‐positive serum samples from MFS/GBS patients. We show that IVIg inhibits the binding of anti‐GQ1b antibodies to GQ1b, thereby preventing complement activation and subsequent pathophysiological effects in our ex vivo mouse NMJ model. These results provide further support for the hypothesis that anti‐ganglioside antibodies are the pathogenic factors in MFS/GBS and show that this NMJ model provides a suitable system for investigating the therapeutic effects of IVIg in antibody‐mediated neuromuscular diseases.
Introduction
High‐dose intravenous immunoglobulin (IVIg) is an established form of treatment in a wide spectrum of immune‐mediated neuromuscular diseases (Dalakas, 1999; Kazatchkine and Kaveri, 2001). The therapeutic effect of IVIg has been demonstrated in randomized clinical trials in patients with Guillain–Barré syndrome (GBS) (van der Meche and Schmitz, 1992; Plasma Exchange/Sandoglobulin Guillain–Barré Syndrome Trial Group, 1997), chronic inflammatory demyelinating polyneuropathy (van Doorn et al., 1991; Dyck et al., 1994), multifocal motor neuropathy (Van den Berg et al., 1995), myasthenia gravis (Gajdos et al., 1997), Lambert–Eaton myasthenic syndrome (Bain et al., 1996), dermatomyositis (Dalakas et al., 1993), stiff‐person syndrome (Dalakas et al., 2001) and in case studies of patients with Miller Fisher syndrome (MFS) (Arakawa et al., 1993; Zifko et al., 1994; Turner and Wills, 2000) and polyneuritis cranialis (Silbert et al., 1992; Wakamoto et al., 2001). A common denominator of these diseases is that they are viewed as being caused by pathogenic autoantibodies directed to well‐defined neuromuscular structures.
The mechanisms by which IVIg exerts these beneficial clinical effects remain unclear, in spite of a number of investigations in in vitro and in vivo systems (Dalakas, 1999; Kazatchkine and Kaveri, 2001). IVIg contains many constituents including immunoglobulin (Ig) G, cytokines and other soluble factors which have pleiotropic and not fully characterized immune‐modulating effects.
Antibodies to the ganglioside GQ1b are strongly associated with the presence of ophthalmoplegia in MFS/GBS patients (Chiba et al., 1993; Willison et al., 1993; Yuki et al., 1993). This is presumably due to the comparatively high concentration of GQ1b in human oculomotor nerves (Chiba et al., 1993, 1997). Ex vivo experiments on mouse diaphragm nerve–muscle preparations have suggested that the neuromuscular junction (NMJ) might be a target of the immune attack in MFS and GBS (Roberts et al., 1994; Willison et al., 1996; Buchwald et al., 1998; Goodyear et al., 1999; Plomp et al., 1999; Bullens et al., 2000; Jacobs et al., 2002a). Our previous ex vivo studies at mouse NMJs have shown that anti‐GQ1b‐positive MFS serum, purified IgG and mouse and human anti‐GQ1b monoclonal antibodies induce a dramatic increase in spontaneous quantal acetylcholine (ACh) release, measured as miniature end‐plate potential (MEPP) frequency, and subsequent blockade of neuromuscular synaptic transmission, due to a failure of ACh release upon nerve impulses (Goodyear et al., 1999; Plomp et al., 1999; Bullens et al., 2000; Jacobs et al., 2002a). Concomitant immunohistological and ultrastructural damage of nerve terminals was also observed (O’Hanlon et al., 2001). These effects were termed the ‘α‐latrotoxin (α‐LTx)‐like effect’, since this spider excitotoxin induces similar pathophysiological effects at the motor nerve terminal (Plomp et al., 1999; O’Hanlon et al., 2001). Superimposition of MEPPs triggered muscle action potentials which induced twitching of muscle fibres (Plomp et al., 1999), a phenomenon on which we have based an efficient bioassay method to screen sera for the ability to induce α‐LTx‐like effects (Jacobs et al., 2002a). Using this ‘twitch bioassay’, we have demonstrated that the α‐LTx‐like effect is specific for anti‐GQ1b antibodies. Twitching is not obvious, however, in MFS/GBS patients, which might be due to smaller MEPP amplitudes and/or action potential firing thresholds in humans, as discussed more extensively in a previous paper (Plomp et al., 1999), but this does not exclude the possibility that the α‐LTx‐like effect and ACh depletion at nerve terminals still occur in these patients and contribute to the craniobulbar weakness. The α‐LTx‐like effect is strictly complement dependent (Goodyear et al., 1999; Plomp et al., 1999; Bullens et al., 2000; O’Hanlon et al., 2001; Jacobs et al., 2002a) and differs in this respect from effects seen by others (Buchwald et al., 1998). In a recent study using genetically modified mice lacking complex gangliosides, we proved that GQ1b, or a closely related complex ganglioside, forms the primary antigenic target of anti‐GQ1b antibodies at the NMJ, instead of some alternative nerve terminal structure (Bullens et al., 2002). Taken together, these results provide strong evidence that anti‐GQ1b antibodies are important pathogenic factors and may act, at least in part, through effects at NMJs.
It was recently shown, in a different model system for analysing deleterious effects of GBS sera on NMJ function, that IVIg partially inhibits a complement‐independent block of ACh release induced by anti‐ganglioside antibody‐ negative and ‐positive GBS sera or purified IgG (Buchwald et al., 2002). In that study, two low‐titre anti‐GQ1b‐positive sera were included and the effects of added IVIg and the purified F(ab)2 portion of IVIg–IgG was studied with the purified IgG from one of the cases. It showed that the ACh release blocking effect of this purified IgG could be inhibited by IVIg and its F(ab)2 portion, indicating that one action of IVIg on anti‐GQ1b antibodies may be anti‐idiotypic neutralization.
In the present study, using our recently developed twitch bioassay in conjunction with standard NMJ microelectrode physiology, immunohistological and immunoassay techniques, we investigated whether and how IVIg might interfere with the complement‐dependent pathophysiological effects of high‐titre anti‐GQ1b‐positive sera at motor nerve terminals. Preliminary results of our studies and those of Buchwald and colleagues were reported at the Peripheral Nerve Society meeting in Telfs, Austria, in September 2001 (Willison et al., 2002). Furthermore, we presented our work in abstract format at the World Muscle Society meeting in Rotterdam, The Netherlands, in October 2002 (Jacobs et al., 2002b).
Material and methods
Patient serum samples
Serum samples with anti‐GQ1b IgG antibodies were obtained before treatment from 12 patients with MFS and two patients with GBS. The MFS patients were clinically typical and suffered from areflexia, ophthalmoplegia and ataxia. The GBS patients fulfilled the criteria for GBS of the National Institute of Neurologic Communicative Disorder and Stroke and were unable to walk 10 m independently. All samples were tested in enzyme‐linked immunosorbent assay (ELISA) studies to investigate the inhibitory effects of IVIg on anti‐GQ1b antibody binding to GQ1b. In studies investigating the effects of anti‐GQ1b antibodies on ex vivo NMJ preparations, serum samples from two randomly selected MFS patients (MFS1 and MFS2) and two GBS patients (GBS1 and GBS2) were used. These two GBS patients and one MFS patient (MFS2) were treated with high‐dose IVIg at a dose of 2 g per kilogram bodyweight and the other MFS patient (MFS1) was treated with plasma exchange. In these cases, serum samples were obtained within 2 weeks of onset of weakness and at different time points during follow‐up. As a source of complement, normal human serum (NHS) was obtained in bulk and frozen in 1 ml aliquots. The serum samples from the patients and the NHS were stored at –70°C until use.
IVIg solution and application
Therapeutic IVIg was either Gammagard S/D (Baxter, Hyland division, Lessines, Belgium), dissolved in sterile water according to the manufacturer’s instructions, or Octagam (Octapharma, Lachen, Switzerland) in premixed solution (5 g/100 ml). To investigate the potential mechanism of action of IVIg on the binding and α‐LTx‐like effects of anti‐GQ1b antibodies, several incubation protocols were developed and studied (Fig. 1). For ex vivo studies on the NMJ, dilution series of Gammagard, dialysed in Ringer medium containing 116 mM NaCl, 4.5 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 1 mM NaH2PO4, 23 mM NaHCO3 and 11 mM glucose, pH 7.4, in a Slide‐A‐Lyzer (Pierce, Rockford, IL) at 4–8°C for at least 15 h, or bovine serum albumin (BSA; Sigma, Zwijndrecht, The Netherlands) as a control protein, were pre‐incubated with serum dilutions in Ringer medium. These mixtures, with a final concentration of IVIg or BSA of ≤25 mg/ml and serum dilutions of ≥1: 2, as stated, were co‐incubated at 32°C for at least 2 h before bath application. Thereafter, the mixtures were applied in the twitch bioassay or in electrophysiological experiments in mouse hemidiaphragm as described below. The majority of ELISA experiments were performed using Octagam, with human serum albumin (HSA; Sigma, Poole, UK) as a control protein, both diluted in phosphate‐buffered saline (PBS, pH 7.4) in dilution series of ≤25 mg/ml. In control studies for ELISAs, Octagam and Gammagard were directly compared in both manufacturer’s stabilizing buffer and when dialysed against PBS.
Anti‐ganglioside antibody ELISA studies
Serum samples were tested initially at 1: 100 dilutions for IgM and IgG antibodies to GM1, GM2, GD1a, GD1b, GD3, GT1a, GT1b and GQ1b by ELISA, and titres were calculated by doubling dilution and end‐point dilution analysis according to methods described previously (Jacobs et al., 1996). In most cases, reactivity with GQ1b was confirmed using thin‐layer chromatography overlay (Jacobs et al., 1996).
Serum samples from all MFS/GBS patients, including the four samples (MFS1, MFS2, GBS1, GBS2) used in the bioassays, were screened in a competitive ELISA for the ability of IVIg to inhibit the binding of anti‐GQ1b to GQ1b. MFS/GBS serum samples diluted 1: 50 were admixed with an equal volume of IVIg or HSA (resulting in a concentration range of 25, 5, 1, 0.2 and 0.04 mg/ml), incubated at room temperature for 1 h and applied to ELISA wells at 4°C for 4 h (Fig. 1, protocol C). ELISA wells were then washed three times with PBS containing 0.005% Tween 20. This was established in pilot studies as the minimum necessary Tween 20 percentage to diminish the non‐specific background signal resulting from the high concentrations of polyclonal human IgG, especially seen in wells exposed to 25 mg/ml IVIg. In all other washing steps, Tween 20 was avoided, as detergent strips ganglioside from ELISA wells, rendering assays highly insensitive. Assays then proceeded as standard, using peroxidase‐conjugated goat anti‐human IgG as secondary antibody and development with substrate.
In ELISAs to determine whether IVIg could displace anti‐GQ1b antibodies once bound to GQ1b, serum samples from two MFS patients (MFS1 and MFS3) were diluted 1: 50 and incubated at 4°C overnight, the plates washed three times in PBS and then exposed to the above concentration range of IVIg or HSA at 4°C for 1 h (Fig. 1, protocol D). Plates were then washed three times with PBS containing 0.005% Tween 20, exposed to secondary antibody and the assay proceeded as standard. In experiments to determine whether IVIg could displace GQ1b from the ELISA plate, plates were incubated with IVIg and washed before MFS sera was added and the standard assay resumed (Fig. 1, protocol B).
C1q ELISA
In order to determine whether the amount of complement factor C1q bound to anti‐GQ1b IgG was affected by IVIg treatment, a C1q capture ELISA was developed. Plates were coated with GQ1b and exposed to MFS/GBS sera diluted 1: 50 at 4°C overnight as described above. Purified human C1q (5 µg/ml; Biogenesis, Poole, UK) was added at room temperature for 2 h and then washed, followed by 1: 100 diluted polyclonal sheep peroxidase‐conjugated anti‐human C1q antibody (Biogenesis). In inhibition assays, IVIg or HSA at 25 and 5 mg/ml was added at room temperature for 1 h, after the C1q incubation, followed by the anti‐human C1q antibody as above.
Twitch bioassay
The capacity of sera to induce muscle fibre twitching, indicating the α‐LTx‐like effect, was tested in the twitch bioassay as described previously (Jacobs et al., 2002a). Briefly, serum samples were incubated at 56°C for 30 min to inactivate complement and diluted 1: 2 and dialysed in Ringer medium at 4–8°C for at least 15 h. Swiss outbred mice (3–4 weeks old) were killed by CO2 inhalation, according to the Leiden University Medical Centre guidelines. Hemidia phragms were longitudinally sectioned in four equally sized strips and pinned out in incubation wells. Strips were incubated in duplicate with 200 µl of complement‐inactivated serum samples diluted in Ringer medium, as stated, at 32°C for 2 h and additionally at 4–8°C for 30 min. Strips not showing muscle contraction upon direct electrical stimulation were discarded. Strips were then incubated at room temperature with NHS, diluted 1: 2 and dialysed in Ringer medium, as a complement source and observed at 40× magnitude using an Olympus SZ40 stereomicroscope to determine the grade of twitching. IVIg (25 mg/ml) was added either before the MFS/GBS serum incubation (Fig. 1, protocol B), during the serum incubation (Fig. 1, protocol C), just before and during the subsequent complement incubation (Fig. 1, protocol E) or only during the complement incubation (Fig. 1, protocol F). Twitching was defined as a fast repetitive asynchronous contraction of muscle fibres (1–5 Hz). Four sectors were discriminated using the horizontal and vertical middle of the strip to grade the twitching as 0 (no contraction), 1 (twitching in one sector), 2 (twitching in two sectors) and 3 (twitching in three or four sectors). A positive serum sample for inducing the α‐LTx‐like effect was defined as having a score of 2 or 3 in at least one of the two duplicate strips within 30 min of observation. Thereafter, the strips were rinsed, snap frozen and stored at –80°C.
We have previously described that α‐LTx‐like effects of anti‐GQ1b antibody and complement at NMJs result in twitching of diaphragm muscle fibres due to action potentials triggered by summated MEPPs (Plomp et al., 1999). In the same study, with control experiments using (+)‐tubocurarine, a blocker of ACh receptors, we excluded the possibility that this twitching was due to malfunction of the muscle fibre membrane. These control experiments were repeated here for the twitch bioassay. Mouse diaphragm strips were pre‐incubated with 50 µg/ml of a prototype anti‐GQ1b mouse monoclonal IgM antibody termed CGM3 (Goodyear et al., 1999) and were subsequently incubated with NHS (1: 2) as complement source. The addition of 2.5 µM (+)‐tubocurarine (Sigma, Zwijndrecht, The Netherlands) to the strips 10 min before and during the 1 h NHS incubation prevented the twitches that were readily observed during the observation period in the positive controls without added (+)‐tubocurarine (a twitch score of 0 in all four tested strips treated with (+)‐tubocurarine, versus twitch scores of 3, i.e. the maximal possible score, in all three tested control strips). This observation confirms that NMJ dysfunction, instead of muscle fibre membrane dysfunction, underlies twitching in the twitch bioassay.
Ex vivo electrophysiological studies
Within hemidiaphragm preparations, muscle fibres were impaled near the NMJ, with a 10–20 MΩ glass microelectrode filled with 3 M KCl. Intracellular recordings of MEPPs were made at 20–22°C using standard recording equipment (Plomp et al., 1999). Signals were digitized and further analysed off‐line. Complement‐inactivated serum samples were diluted 1: 2 and dialysed with Ringer medium. Hemidiaphragms were pinned out on silicone rubber and incubated in a closed vial in 1.5 ml of MFS/GBS sera diluted in Ringer medium, as stated, at 32°C for 2–2.5 h, followed by incubation at 4–8°C for 45 min. After incubation, the preparation was placed in Ringer medium in a 2 ml recording bath and re‐warmed to 20°C, and at least 30 MEPPs were measured at 3–7 NMJs during a 20 min recording period. Subsequently, the Ringer medium was replaced with NHS, and MEPPs were measured at 7–15 NMJs during a 45 min recording period at 20°C. IVIg (25 mg/ml) was added either during the MFS/GBS serum incubation (Fig. 1, protocol C) or just before and during the subsequent complement incubation (Fig. 1, protocol E). Control measurements were taken at NMJs of contralateral hemidiaphragms in medium that contained no IVIg or BSA as protein control.
Immunostaining
Muscle strips observed in the twitch bioassay were defrosted, fixed with 2% paraformaldehyde in PBS at 4°C for 2 h and prepared for immunohistological analysis as described previously (Goodyear et al., 1999; Plomp et al., 1999). Briefly, longitudinal 10 µm sections were co‐stained with 1: 2000 dilution (0.5 µg/ml) of Texas Red‐conjugated α‐bungarotoxin (Molecule Probes, Leiden, The Netherlands), a ligand for ACh receptors, and with 1: 200 fluorescein isothiocyanate‐conjugated anti‐C3c antibodies (Dako, Glostrup, Denmark) to detect deposition of activated human complement, in PBS and 0.1% Triton TX at 4°C for 1 h. The slides were rinsed four times in 4°C PBS and then mounted in Citifluor antifade (Citifluor Products, Canterbury, UK) and stored at –20°C.
Imaging and quantification of immunostaining
Digital images were obtained either by means of a Sony colour CCD camera mounted on a Zeiss Axioplan fluorescence microscope (magnitude 100×) and linked to an image archiving system (Sirrius VI; Optivision) or by a Zeiss Pascal confocal microscope. Bitmap processing and annotation were conducted on Photo Magic and Windows Draw (both by Micrographx). Morphometric measurements were made using Scion Image (Scion, Frederick, MD) or Aequitas IA (Dynamic Data Links, Cambridge UK) image analysis software.
A quantitative measure of C3c deposition, termed the ‘complement load’, expressed the C3c signal as a percentage of the area of an individual end‐plate. Briefly, NMJs were identified by α‐bungarotoxin staining, and images of the α‐bungarotoxin and associated C3c stain were recorded under standardized camera conditions. Aequitas or Scion Image software was used to delineate the area of the α‐bungarotoxin signal of individual end‐plates and to express the overlaying C3c signal as a percentage of this area. The complement loads recorded from individual staining runs were expressed as a percentage of an internal standard. This technique is explained in greater detail elsewhere (O’Hanlon et al., 2001).
As a related parameter, end‐plates were scored as positive or negative for C3c, based on whether or not any signal was detected over the NMJ using the same predetermined camera settings as above. In each specimen, 20 end‐plates were measured, with the observer blinded for the treatment of the strips.
Statistical analysis
The Wilcoxon–Mann‐Whitney U‐test or Student’s t‐test were used as indicated. A P‐value of <0.05 was considered to be significant.
Results
Inhibition of serum antibody binding to ganglioside GQ1b by IVIg
Serum samples from all MFS/GBS patients had high titres of anti‐GQ1b IgG antibodies (range 1: 1000 to 1: 100 000). Titres for the four samples used in the ex vivo bioassay (MFS1, MFS2, GBS1 and GBS2) are shown in Table 1. MFS2 also had a low titre (1: 100) of anti‐GQ1b IgM antibodies. The anti‐GQ1b IgG antibodies were all of the IgG1 and/or IgG3 subclass, and all four samples had additional IgG reactivity to GT1a. GBS1 and GBS2 also reacted with GD3. The NHS (diluted 1: 100) used as a complement source contained no detectable levels of anti‐GQ1b antibodies.
Using the panel of 12 MFS and two GBS sera, the inhibitory effect of IVIg on serum antibody binding to GQ1b was studied in ELISAs according to protocol C (Fig. 1), and results of the pooled data from all sera are shown in Fig. 2. HSA exhibited a mild but significant inhibitory effect at all doses. The 14 sera exhibited a dose‐dependent inhibition of binding to GQ1b by IVIg that differed significantly from the HSA effect at 1, 5 and 25 mg/ml.
Neither IVIg preparations (25 mg/ml) showed direct binding to GQ1b, displacement of GQ1b from ELISA plates (Fig. 1, protocol B) or inhibition of secondary antibody activity (Fig. 1, protocol F; data not shown).
α‐LTx‐like effects of MFS/GBS sera at mouse NMJs
First, the baseline effects of the four anti‐GQ1b antibody‐containing sera (MFS1, MFS2, GBS1 and GBS2) were determined as standard (Fig. 1, protocol A). Samples were tested at titres ranging from 1: 2 to 1: 480 and induced twitching in the twitch bioassay that started within 5 min of adding NHS as the complement source and continued for at least 20 min. In microelectrode studies conducted for comparison, all samples induced at least a 13‐fold increase in MEPP frequency after the addition of the complement source (Table 1). The mean MEPP frequencies before serum application, after MFS/GBS serum incubation and after complement incubation were 0.39 ± 0.05, 0.37 ± 0.07 and 32.43 ± 10.54/s, respectively (n = 4, mean ± SEM). Incubation with NHS alone induced no twitching or increase in MEPP frequency.
Inhibiting action of IVIg on the α‐LTx‐like effects of MFS/GBS sera at mouse NMJs
The twitch assay was used to screen and quantitate the IVIg effects, using dilution series of sera and IVIg in several different incubation protocols. Serum samples from each of the four MFS/GBS patients were pre‐incubated with IVIg (25 mg/ml) for 2 h, and this mixture was then incubated in the twitch bioassay, followed by NHS as a complement source (Fig. 1, protocol C). This prevented twitching with all samples (twitch score of 0 in all samples). In this series of studies, the minimal concentration of IVIg required to prevent twitching was determined by dilution analysis of IVIg over the range 1–25 mg/ml, and it was shown to be 25 mg/ml for MFS1, 15 mg/ml for MFS2, 5 mg/ml for GBS1 and 25 mg/ml for GBS2. BSA (25 mg/ml) as a protein control, mixed with each of the four serum samples, did not prevent twitching (Table 2).
In ex vivo microelectrode electrophysiology, we confirmed the inhibitory action of IVIg seen in the twitch bioassay by recording MEPPs at NMJs of hemidiaphragm preparations that were treated with MFS/GBS sera, complement and IVIg using the same incubation protocol C (Fig. 1). When the hemidiaphragm was pre‐incubated with MFS/GBS sera containing IVIg (25 mg/ml), with subsequent addition of NHS as a complement source, the increase in MEPP frequency was greatly inhibited with sera MFS1, MFS2 and GBS1 and, to a lesser extent, with GBS2 (Fig. 3A). BSA mixed with these serum samples had no inhibitory effect.
Incubation with IVIg or BSA (25 mg/ml) alone, followed by incubation with NHS (1: 2), induced no twitching or increase in MEPP frequency (data not shown).
Analysis of the mechanism of the protective effect of IVIg
The above results demonstrate that admixture of MFS sera with IVIg at the step when muscle preparations are incubated with MFS/GBS sera (Fig. 1, protocol C) abrogates the anti‐GQ1b antibody‐mediated effects at the nerve terminal. Possible explanations are that (i) nerve terminal GQ1b is being masked or removed by IVIg; (ii) anti‐GQ1b antibody is not being deposited to nerve terminals (and is therefore not available to fix complement); or (iii) complement fixation by tissue‐bound antibody is being inhibited. We were unable to address point (ii) directly because we could not quantify MFS/GBS anti‐GQ1b IgG at nerve terminals in these experiments: these very low IgG levels only just reached the detection threshold of our imaging system and were further obscured by non‐specific deposition of human IgG present in the high levels of therapeutic IVIg.
In order to address point (i), we pre‐incubated the diaphragm preparation with IVIg (25 mg/ml) for 2 h, removed the excess IVIg, and added anti‐GQ1b‐containing serum and complement (Fig. 1, protocol B). This did not prevent the induction of twitching by the MFS/GBS sera, indicating that GQ1b is not removed or otherwise obscured by IVIg (data not shown). Similarly, in ELISAs, this pre‐incubation step had no effect on the anti‐GQ1b antibody signal (data not shown).
With respect to point (iii), and to address a therapeutically more relevant situation, we incubated MFS/GBS sera in the twitch assay and then added the NHS, as a complement source, admixed with IVIg (25 mg/ml) (Fig. 1, protocol F). In this situation, twitching was not prevented (Table 2), indicating that complement activation was not being inhibited. However, this also raised the possibility that the complement‐mediated effect was able to proceed at a faster rate than the onset of any inhibitory effect of IVIg. To examine this, we incubated muscle strips in the twitch assay with MFS/GBS sera, exposed them to IVIg for 1 h and added the IVIg–complement mixture (Fig. 1, protocol E); in this situation, twitching was abolished in three out of the four samples (MFS1, MFS2 and GBS2, but not GBS1).
The effect of IVIg on the α‐LTx‐like effect in protocol E (Fig. 1) was further quantified in the twitch bioassay using GBS2 serum, since this serum contains a very high titre of anti‐GQ1b antibodies (>1: 100,000) and was relatively resistant to the blocking effect of IVIg at low serum dilutions. We conducted a more detailed dilution analysis of the twitching effect of GBS2 serum, at a fixed concentration of IVIg or BSA (25 mg/ml). In this analysis, GBS2 serum induced twitching at dilutions up to 1: 480 that was unaffected by BSA (Fig. 4A). Complete inhibition of twitching by IVIg was reached at GBS2 dilutions over 1: 200 (Fig. 4A). To confirm this result electrophysiologically, we exposed GBS2 serum (diluted 1: 31) pre‐incubated hemidiaphragm preparations to IVIg for 1 h and then added the IVIg–complement mixture (Fig. 1, protocol E), and we observed a greatly reduced α‐LTx‐like effect (Fig. 3B). Thus, the difference in results observed between protocols E and F indicates that the protective effect of IVIg requires a short time window in which to act before the addition of a complement source.
Inhibition of complement deposition at mouse NMJs by IVIg
The protective effects of IVIg could be due to the prevention of complement deposition at nerve terminals. In order to investigate this, diaphragm strips from the twitch bioassay were co‐stained with anti‐C3c antibodies and α‐bungarotoxin to quantify the deposition of complement activation marker C3c. Strips incubated with sera from the four MFS/GBS patients all showed significant C3c depositions at end‐plates, ranging from 45 to 75% (mean 59%) of end‐plates (Table 2). MFS/GBS serum samples simultaneously incubated with 25 mg/ml of IVIg (Fig. 1, protocol C) reduced the number of C3c‐positive end‐plates to between 0 and 10% (mean 4%; P = 0.02, as shown in Table 2). No reduction was observed when the serum samples were mixed with the same concentrations of BSA. When IVIg (25 mg/ml) was co‐incubated with the complement source (Fig. 1, protocol F), the percentage of C3c‐positive end‐plates was not significantly altered (35–65%, mean 46%). In control samples, IVIg and NHS treatment alone (in the absence of MFS/GBS sera) induced detectable C3c depositions at 7 and 4% of end‐plates, respectively.
In the series of studies titrating the effects of GBS2 serum dilution on the α‐LTx‐like effect, IVIg was added for 1 h before and during the incubation with the NHS–IVIg mixture. In this paradigm (Fig. 1, protocol E), a reduction in the number of C3c‐positive end‐plates was observed, to 23–85% of the BSA control value (Fig. 4B). The largest reduction, to 23% of the BSA control, was found at GBS2 dilutions of 1: 240, the dilution at which the twitch score approached 0 in the dilution curve (Fig. 4A). The relative C3c load at end‐plates was also quantitated in this experiment and showed a large and significant reduction compared with BSA at all serum dilutions (Fig. 4C). Typical images used in this analysis are shown in Fig. 5. Other illustrative images of C3c‐stained NMJs appear in previous publications (Plomp et al., 1999; O’Hanlon et al., 2002).
At the higher GBS2 serum dilutions, BSA also had a reducing effect on C3c load at NMJs. However, no reduction of twitch bioassay score was observed. The twitch assay is a very sensitive test for the occurrence of the α‐LTx‐like effect at NMJs, as we have discussed previously (Jacobs et al., 2002a). If only a few NMJs in a strip display the α‐LTx‐like effect, the strip will already score positive. Apparently, much more complement is deposited on average at NMJs than minimally needed to score maximally in the twitch bioassay. From the IVIg curves in Fig. 4A and B, it can be deduced that the percentage of C3c‐positive NMJs must be lower than ∼20% to result in significant inhibition of the twitch score. The ∼30% C3c‐positive end‐plates at the highest dilution of GBS2 in the BSA control group was still enough to score maximally in the twitch bioassay. Extrapolation of the BSA C3c curves in Fig. 4B and C suggests that a further dilution step of GBS2 would presumably have resulted in significantly reduced twitch score by BSA.
Displacement of anti‐GQ1b antibody from GQ1b reduces the amount of antibody available for activating complement
We next determined whether IVIg might interfere with the ability of anti‐GQ1b antibodies to activate complement. Using a C1q ELISA, we first demonstrated that MFS/GBS anti‐GQ1b antibodies bound overnight to GQ1b in ELISA wells were subsequently able to fix C1q (data not shown). When IVIg (5 and 25 mg/ml) was added to ELISA wells for 1 h after incubation of GQ1b plates with MFS/GBS sera but before the addition of C1q, the C1q signal was diminished by 45–55%, indicating interference by IVIg. HSA had no reducing effect. We considered that this might be due in part to competitive displacement of anti‐GQ1b antibodies from GQ1b, rather than the inability of anti‐GQ1b antibodies to fix C1q in the presence of IVIg. To investigate this, we incubated GQ1b‐coated ELISA plates with MFS1 and MFS3 sera (anti‐GQ1b IgG titre of 1: 1200) for 16 h to allow capture of the maximal amount of anti‐GQ1b antibody, discarded excess MFS serum and then added IVIg for 1 h, followed by secondary antibody (Fig. 1, protocol D). Surprisingly, IVIg was able to displace MFS‐associated anti‐GQ1b antibodies bound to GQ1b in a dose‐dependent manner with both sera, and this effect was seen with both Octagam and Gammagard IVIg preparations, both in manufacturer’s diluent (Fig. 6) and when dialysed against PBS (data not shown).
α‐LTx‐like activity in convalescent serum samples from MFS/GBS patients
The capacity of MFS/GBS follow‐up samples obtained after IVIg therapy to induce α‐LTx‐like activity at NMJs was also tested. Anti‐GQ1b IgG antibodies were present in all follow‐up serum samples until at least 4 weeks after IVIg therapy. Convalescence sera from MFS2 and GBS1, obtained ≥2 weeks after IVIg therapy and diluted 1: 2 in Ringer solution, did not induce twitching, an increase in MEPP frequency or significant C3c deposition at end‐plates in the twitch bioassay (Table 3). Convalescence serum from GBS2, obtained 2 weeks after IVIg therapy and diluted 1: 2, induced twitching and an increase in MEPP frequency, whereas a sample obtained 26 weeks after IVIg therapy did not induce these effects or C3c depositions at end‐plates.
Discussion
In this study, we have demonstrated that IVIg inhibits the whole cascade of pathogenic effects of anti‐GQ1b‐positive sera at mouse NMJs ex vivo. In ELISAs, IVIg reduced anti‐GQ1b antibody binding to GQ1b. These inhibiting effects may explain the therapeutic effect of IVIg in MFS/GBS patients with these specific antibodies and are in broad agreement with another recent study on inhibiting effects of IVIg on experimental pathophysiological effects of anti‐ganglioside antibodies at NMJs (Buchwald et al., 2002).
IVIg inhibits binding of serum antibodies to GQ1b
Our data show that IVIg dose‐dependently reduces the serum antibody activity to GQ1b in ELISAs. This effect could be caused by displacement or sheltering of GQ1b ganglioside, by inhibition of the binding of second step antibodies or by immune complex forming with serum anti‐GQ1b antibodies. We experimentally excluded the first two possibilities as far as possible. It remains possible that IVIg contains non‐complement fixing antibodies of low avidity that bind to GQ1b and prevent binding of serum anti‐GQ1b antibodies with higher avidity and complement fixing properties. However, anti‐GQ1b IgG activity is invariably undetectable in serum from healthy controls and could not been demonstrated in therapeutic levels of IVIg (25 mg/ml).
Another possibility is that IVIg directly neutralizes the serum antibodies by exposing GQ1b‐like structures on IgG molecules within IVIg that compete with the GQ1b on the ELISA plates. Serum anti‐GQ1b antibodies in MFS/GBS patients usually cross‐react with GT1a and half of them with GD1b or GD3, indicating that they recognize the common NeuAc(α2–8)NeuAc(α2–3)Gal disialosyl moiety (Chiba et al., 1993; Willison et al., 1993; Yuki et al., 1993). Serum anti‐ganglioside antibodies may be captured by binding to carbohydrate epitopes on IgG, as has been demonstrated for anti‐GM1 antibodies (Thomas et al., 1989). However, the disialosyl moiety recognized by anti‐GQ1b antibodies has so far not been demonstrated on human IgG.
It is more likely that IVIg expresses non‐carbohydrate tertiary structures which interact with the antigen‐binding sites of the anti‐GQ1b antibodies. IVIg preparations contain a wide range of anti‐idiotypic antibodies that recognize the variable regions of autoantibodies and hence may neutralize these antibodies, thereby preventing them from binding to their target autoantigens (Dalakas, 1999; Kazatchkine and Kaveri, 2001). In vitro, non‐carbohydrate epitopes in IVIg preparations have been shown to inhibit the binding of cholera toxin and serum anti‐GM1 antibodies from neuropathy patients to GM1 (Malik et al., 1996; Yuki and Miyagi, 1996; Lopez et al., 2000). More importantly, anti‐idiotypic mouse IgG antibodies have been demonstrated to carry an internal image of GD3, which shares this carbohydrate moiety with GQ1b/GT1a (Chapman and Houghton, 1991). Furthermore, anti‐idiotypic neutralization of anti‐GQ1b, anti‐GM1 and unknown GBS antibodies by IVIg has been demonstrated in a study on the inhibiting effects of IVIg on the complement‐independent blocking effects of GBS sera/IgGs on ACh release at NMJs, using a perfused macro‐patch‐clamp configuration (Buchwald et al., 2002). Our present finding that IVIg directly interferes with the anti‐GQ1b antibodies in the NMJ model studied here, as well as in our ELISAs, is in good agreement with the results of that study.
The presence of dimeric antibody complexes in IVIg preparations has been directly visualized with electron microscopy. Such complexes are absent in the IgG preparation from a single donor (Tankersley et al., 1988). Interestingly, the percentage of dimers in IVIg preparations is associated with its immunosuppressive effects (Teeling et al., 2001). Anti‐idiotypic antibodies, therefore, seem to play an important role in the therapeutic effect of IVIg in antibody‐mediated diseases.
Our study shows that IVIg not only can prevent the binding of anti‐GQ1b antibodies, but also can displace antibodies already bound to GQ1b in ELISAs. This contrasts with other studies, although done on anti‐GM1 antibodies with a different reporting system (Lopez et al., 2000).
In addition to acute neutralization and displacement of anti‐GQ1b/GT1a antibodies, IVIg may have additional, long‐term effects which reduce the level of serum autoantibodies. IVIg also leads to increased catabolism of serum IgG, including that of autoantibodies, and to inhibition of antibody production by a downregulation of B cells (Dalakas, 1999; Yu and Lennon, 1999). These long‐term effects of IVIg are difficult to evaluate in monophasic diseases such as MFS and GBS and cannot explain the rapid clinical improvement sometimes observed in these patients after IVIg therapy.
There are indications that the F(ab)2 portion of IVIg–IgG, rather than the Fc portion, is responsible for neutralization action on pathogenic antibodies (Dietrich and Kazatchkine, 1990; Kazatchkine et al., 1994; Kaveri et al., 1997; Buchwald et al., 2002). As yet, we have not investigated possible differential actions of different IVIg–IgG regions on the effects of anti‐GQ1b‐positive sera in our NMJ model system and ELISAs. However, in view of the studies mentioned above, it is likely that the F(ab)2 portion is responsible for the inhibitory actions of IVIg on anti‐GQ1b antibodies found in the present study.
GBS2 serum was relatively resistant to the inhibiting effect of IVIg at low serum dilutions. This was presumably due to the extremely high anti‐GQ1b antibody titre in this serum, compared with that of MFS1, MFS2 and GBS1 sera. However, within these three lower‐titre sera, there was no clear correlation between titre and inhibiting effects of IVIg. In this respect, it is of interest to note that, in the studies of Buchwald et al. (2002), there is also no clear correlation between anti‐GM1 titre of sera and the effect of IVIg treatment. Apparently, IVIg concentrations used in that study and in our present studies were supramaximal. Furthermore, the fine specificity of antibody for closely related ganglioside antigens and antibody–antigen affinity/avidity may be more significant factors than antibody titre in determining the pathophysiological actions of sera.
IVIg inhibits complement activation by anti‐GQ1b antibodies
The induction of the α‐LTx‐like effect at mouse NMJs by anti‐GQ1b antibodies is strictly complement dependent (Goodyear et al., 1999; Plomp et al., 1999; Bullens et al., 2000; O’Hanlon et al., 2001; Jacobs et al., 2002a). The complement activation marker C3d has been shown to reflect the localization of the peripheral nerve damage in GBS patients, i.e. the axolemma of motor fibres in patients with acute motor axonal neuropathy and the myelin in patients with acute inflammatory demyelinating polyneuropathy, suggesting that complement activation is presumably mediating nerve damage (Hafer‐Macko et al., 1996a, b).
Our study shows that IVIg reduces complement binding by anti‐GQ1b antibodies in vitro and complement activation in the mouse diaphragm model ex vivo. It is uncertain whether this is due to displacement of anti‐GQ1b antibody from GQ1b, rather than to a direct effect on C1q binding or on other activated complement factors. In ELISAs, clinically relevant concentrations of IVIg dose‐dependently reduced the C1q binding, but also reduced the amount of IgG antibodies attached to GQ1b. On the basis of our present experiments, it is therefore not possible to conclude whether IVIg has a direct inhibiting effect on C1q complex formation. Inhibition of C1q binding by IVIg was previously demonstrated for antibody‐sensitized erythrocytes (Wada et al., 2001), although this was not confirmed by others (Basta et al., 1991). This mechanism of IVIg does not necessarily contribute to inhibition of the α‐LTx‐like effect, since this effect may be able to proceed independently of C1q through activation of the alternative pathway (Plomp et al., 1999). Simultaneous incubation of IVIg and patient serum prevented the α‐LTx‐like effect and reduced the deposition of the complement at mouse NMJs. This inhibitory effect did not occur when IVIg was added to the mouse diaphragm strips, and the excess rinsed away, before MFS/GBS serum incubation. Inhibition of complement activation by IVIg in the ex vivo assays could be attributed to neutralization and displacement of anti‐GQ1b antibodies as seen in ELISAs, since adding IVIg after incubation with patient serum induced an inhibitory effect. However, IVIg may also directly interfere with complement activation by inhibiting binding or activation of complement factors by anti‐GQ1b antibodies bound to mouse NMJs. IgG in IVIg preparations binds to complement factors C3, C4, C3a and C5a (Shohet et al., 1993; Wada et al., 2001; Basta et al., 2003) and inhibits the incorporation of the available C3b molecules into the C5 convertase assembly (Basta and Dalakas, 1994). Furthermore, it interferes with the activation of C5 and/or C8. The complement inhibition hypothesis is further supported by findings in dermatomyositis patients, in whom IVIg inhibited the uptake of C3b and C4b and the deposition of membrane attack complex on endomysial capillaries (Basta and Dalakas, 1994; Basta et al., 1996; Dalakas, 1996). Thus, effects of IVIg on complement binding and activation may be relevant for the whole spectrum of antibody and complement‐mediated neuromuscular diseases. However, while our results with protocol E and MFS1, MFS2 and GBS2 sera (see Results and Figs 3 and 4) might be explained by direct inhibition of complement by IVIg, the lack of inhibition of effects using protocol F (Table 2) seems to argue against a major or very rapid inhibitory effect of IVIg on late stages of the complement cascade.
Does IVIg inhibit the pathophysiological effects of anti‐GQ1b antibodies at NMJs of MFS/GBS patients?
The α‐LTx‐like effect is highly associated with anti‐GQ1b antibodies and specific for sera from MFS/GBS patients (Jacobs et al., 2002a), suggesting that the NMJ may be one of the targets in these diseases. Preliminary clinical electrophysiological studies have indeed implicated NMJ dysfunction in paralysis of MFS/GBS patients with antibodies against GQ1b (Uncini and Lugaresi, 1999; Wirguin et al., 2002) or other gangliosides (Ho et al., 1997; Spaans et al., 2003). Many MFS/GBS patients show a clinical improvement, either spontaneously or after IVIg, that is too rapid to be explained by repair of myelin sheaths or axons. Post‐IVIg samples rapidly lost the capacity to induce the α‐LTx‐like effect, although some remaining anti‐GQ1b antibody could still be detected. These results indicate that the ELISA is more sensitive than the bioassay in detecting anti‐GQ1b antibodies and suggest that a minimal titre of anti‐GQ1b antibodies is needed to induce the α‐LTx‐like effect. Alternatively, IVIg in post‐treatment samples might have interfered with the action of serum anti‐GQ1b antibodies in the bioassays.
The blood–nerve barrier is naturally deficient at the NMJ, making this structure easier accessible for both anti‐GQ1b antibodies and IVIg, in comparison with other peripheral nerve sites. It is unclear whether this would lead to a localized effect of IVIg at patient NMJs.
Treatment of patients with high‐dose IVIg of 2 g/kg induces serum IgG levels of 32–50 mg/ml (Sekul et al., 1994), which is in the range in which IVIg induced the inhibitory effects in our study. This indicates that the experimental findings in our study may be directly relevant for understanding the clinical modes of action of IVIg treatment in MFS and a subgroup of GBS patients.
Conclusion
The data in the present study, together with a recent study on inhibiting effects of IVIg in a different experimental NMJ model for the action of MFS/GBS serum antibodies (Buchwald et al., 2002), indicate that IVIg directly interferes with the pathogenic effects of anti‐GQ1b antibodies. This work supplies further evidence in favour of the hypothesis that anti‐ganglioside antibodies are the pathogenic factors in MFS/GBS and gives experimental background to the clinical observation that IVIg application is an effective form of treatment.
Acknowledgements
This research project was sponsored by a European Neurological Society Fellowship Stipend (to B.C.J.) and grants from the Prinses Beatrix Fonds (96‐0210, to R.W.M.B.), KNAW Van Leersumfonds (to J.J.P.) and the Guillain–Barré Support Group of Great Britain and the Wellcome Trust (to H.J.W.).
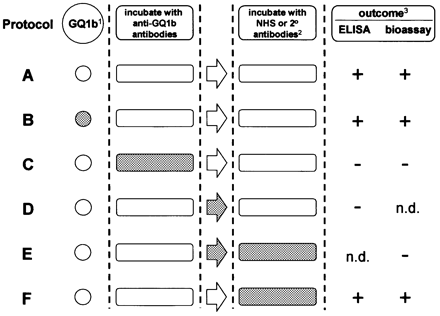
Fig. 1 Schematic summary of the protocol design used to monitor the intravenous immunoglobulin (IVIg) effects. Shading indicates presence of IVIg. 1GQ1b, either coated on enzyme‐linked immunosorbent assay (ELISA) plates or present as a component of the neuromuscular junction in the bioassay. 2Normal human serum (NHS) as a complement source in the bioassay; peroxidase‐labelled goat anti‐human IgG (2°) antibodies in ELISAs. 3Outcome as determined by anti‐GQ1b IgG binding in ELISAs and by twitching, increased frequency of miniature end‐plate potentials or C3c deposition in bioassay (which showed 100% agreement). + indicates that positive outcome was unaffected by IVIg; – indicates that IVIg had an inhibitory effect.
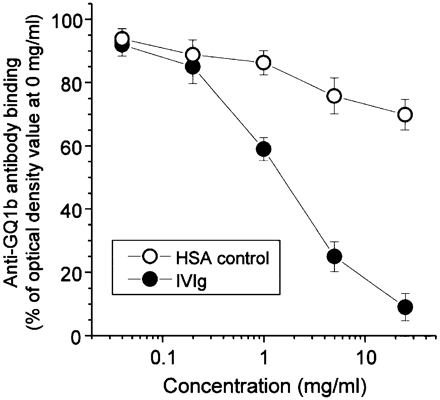
Fig. 2 Inhibition of serum antibody binding to ganglioside GQ1b by intravenous immunoglobulin (IVIg) in enzyme‐linked immunosorbent assay. Serum samples with anti‐GQ1b immunoglobulin G (IgG) antibodies from 12 Miller Fisher syndrome (MFS) and two Guillain–Barré syndrome (GBS) patients were each diluted 1: 50 and mixed with a dilution series of IVIg or human serum albumin (HSA) (Fig. 1, protocol C). The binding of serum anti‐GQ1b antibodies was expressed as the percentage of the optical density of the serum dilution mixed with 0 mg/ml of IVIg or HSA. All optical densities are presented with blank (ganglioside‐free) wells substracted. Dose‐dependent inhibition of the anti‐GQ1b IgG activity by IVIg in sera from 12 MFS and two GBS patients compared with the effect of similar concentrations of HSA (mean values).
![Fig. 3In vitro electrophysiological confirmation of the inhibiting effect of intravenous immunoglobulin (IVIg) on the α‐latrotoxin‐like effects of anti‐GQ1b‐positive Miller Fisher syndrome (MFS) and Guillain–Barré syndrome (GBS) sera at mouse neuromuscular junctions (NMJs). Spontaneous quantal transmitter release [miniature end‐plate potential (MEPP) frequency] was measured at the mouse NMJ in left and right hemidiaphragm muscle–nerve preparations before and after serum incubation and during subsequent addition of normal control serum. (A) IVIg (25 mg/ml), added during MFS/GBS serum incubation (Fig. 1, protocol C), inhibited the increase of MEPP frequency seen in the contralateral hemidiaphragm under control conditions. Serum dilution were as follows: MFS1, 1: 7; MFS2, 1: 2; GBS1, 1: 7; and GBS2, 1: 31. MFS1, MFS2 and GBS1 sera were each tested on two muscles (7–15 NMJs sampled per measuring session); GBS2 serum was tested on one muscle (7–18 NMJs sampled per measuring session). (B) Inhibiting effect of IVIg when added 1 h before and during the phase of the experiment when complement was present (Fig. 1, protocol E). GBS2 serum was tested in this way in four muscles (7–15 NMJs sampled per measuring session). BSA = bovine serum albumin.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/brain/126/10/10.1093/brain/awg235/2/m_awg235f3.gif?Expires=1716470593&Signature=usQTuNTy5Q6uEbcuNQCz3ncZFZMUzPjH5NRJCY3kogj2auby4b5npAN11NeQZq07IQP~5DtLgywzrqZSw2EZwdbMKasFKi2O1dY-hefp0FxvPjdtc2V16ccTiybmp18dOInxr5OspWKUsy2Yu-3JGDphA2V26bNfAiYNb0HaBM-VZKa3iSBqUBQZO4Hu1HBwoHMv2b~hZUNQrVvGtHZl-SEr2HqtRyPkSEuhsR5HQ97F42h3hSJRnwalv~YwU8JWpdXSEaPQo60-~hXugx4~A2~wmwKIH3KdD19XqiCWqfQdF2WummfgIhVXtZ3TChfb3QvHra-POVwMqNWzWEBTsw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Fig. 3In vitro electrophysiological confirmation of the inhibiting effect of intravenous immunoglobulin (IVIg) on the α‐latrotoxin‐like effects of anti‐GQ1b‐positive Miller Fisher syndrome (MFS) and Guillain–Barré syndrome (GBS) sera at mouse neuromuscular junctions (NMJs). Spontaneous quantal transmitter release [miniature end‐plate potential (MEPP) frequency] was measured at the mouse NMJ in left and right hemidiaphragm muscle–nerve preparations before and after serum incubation and during subsequent addition of normal control serum. (A) IVIg (25 mg/ml), added during MFS/GBS serum incubation (Fig. 1, protocol C), inhibited the increase of MEPP frequency seen in the contralateral hemidiaphragm under control conditions. Serum dilution were as follows: MFS1, 1: 7; MFS2, 1: 2; GBS1, 1: 7; and GBS2, 1: 31. MFS1, MFS2 and GBS1 sera were each tested on two muscles (7–15 NMJs sampled per measuring session); GBS2 serum was tested on one muscle (7–18 NMJs sampled per measuring session). (B) Inhibiting effect of IVIg when added 1 h before and during the phase of the experiment when complement was present (Fig. 1, protocol E). GBS2 serum was tested in this way in four muscles (7–15 NMJs sampled per measuring session). BSA = bovine serum albumin.
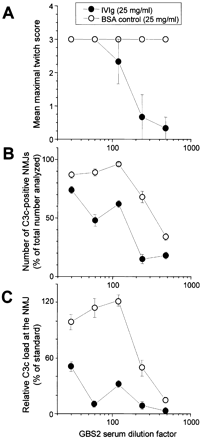
Fig. 4 Inhibition of serum anti‐GQ1b IgG antibody‐mediated twitching of muscle fibres and complement activation at neuromuscular junctions (NMJs) by intravenous immunoglobulin (IVIg). (A) The effects of a dilution series of Guillain–Barré syndrome serum (GBS2) was determined in the twitch bioassay, with IVIg (25 mg/ml), or bovine serum albumin (BSA, 25 mg/ml) as a control, added in the 1 h incubation period before, and during the addition of normal human serum as complement source (Fig. 1, protocol E). Each serum dilution was tested in triplicate. C3c deposition at NMJs was determined in muscle strips from the twitch bioassay experiments and expressed as the number of C3c‐positive NMJs as percentage of the total analysed (n = 1700) (B) and as the relative load of C3c deposited per end‐plate analysed (C).
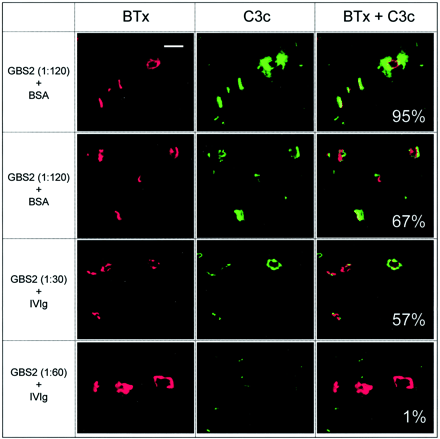
Fig. 5 Typical fluorescent confocal micrographs used for the quantitative analysis of complement load. Examples of pictures obtained in the series of studies titrating the effects of a Guillain–Barré syndrome serum (GBS2) dilution on the α‐latrotoxin‐like effect, in the presence of either bovine serum albumin (BSA; as a control) or intravenous immunoglobulin (IVIg) (Fig. 4). The α‐bungarotoxin signal (BTx), which was used to delineate the neuromuscular junction (NMJ), is shown in the first column, the complement signal (C3c) is shown in the second and the overlaid signals are shown in the third. The figure illustrates that, where present, the complement deposits are largely concentrated at the NMJ. In the third column, the percentage number represents the average complement load per area NMJ for all the end‐plates in the image shown. Scale bar = 50 µm.
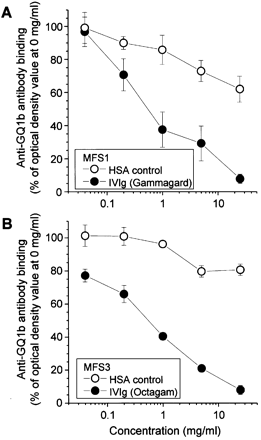
Fig. 6 Displacement of anti‐GQ1b antibodies from GQ1b by intravenous immunoglobulin (IVIg) in enzyme‐linked immunosorbent assay (ELISA). GQ1b‐coated ELISA plates were incubated with Miller Fisher syndrome serum MFS1 (A) or MFS3 (B) overnight to capture anti‐GQ1b antibody, excess MFS serum was discarded and then IVIg or human serum albumin (HSA, serially diluted from 0.025 to 25 mg/ml) was added for 1 h (Fig. 1, protocol D). Plates were then washed, exposed to secondary antibody and developed as standard. IVIg is able to displace anti‐GQ1b antibodies previously bound to GQ1b. The effect is dose dependent and occurs with both IVIg preparations studied (Octagam and Gammagard).
Clinical features, serum anti‐GQ1b antibody titres and α‐latrotoxin‐like effects of four patients with MFS or GBS
| Patient | Ophth | Treatment | Titre anti‐GQ1b IgG | Twitching | ↑fMEPP |
| MFS1 | + | PE | 1: 4800 | + | + |
| MFS2 | + | IVIg | 1: 51200 | + | + |
| GBS1 | + | IVIg | 1: 12800 | + | + |
| GBS2 | + | IVIg | 1: 102400 | + | + |
| Patient | Ophth | Treatment | Titre anti‐GQ1b IgG | Twitching | ↑fMEPP |
| MFS1 | + | PE | 1: 4800 | + | + |
| MFS2 | + | IVIg | 1: 51200 | + | + |
| GBS1 | + | IVIg | 1: 12800 | + | + |
| GBS2 | + | IVIg | 1: 102400 | + | + |
MFS = Miller Fisher syndrome; GBS = Guillain–Barré syndrome; Ophth = ophthalmoplegia; IgG = immunoglobulin G; ↑fMEPP = increased frequency of miniature end‐plate potentials (>1.8/s); serum dilutions ≥1: 100 (titre anti‐GQ1b IgG) or 1: 2 (twitching, fMEPP); + = present. PE = plasma exchange; IVIg = intravenous immunoglobulins.
Clinical features, serum anti‐GQ1b antibody titres and α‐latrotoxin‐like effects of four patients with MFS or GBS
| Patient | Ophth | Treatment | Titre anti‐GQ1b IgG | Twitching | ↑fMEPP |
| MFS1 | + | PE | 1: 4800 | + | + |
| MFS2 | + | IVIg | 1: 51200 | + | + |
| GBS1 | + | IVIg | 1: 12800 | + | + |
| GBS2 | + | IVIg | 1: 102400 | + | + |
| Patient | Ophth | Treatment | Titre anti‐GQ1b IgG | Twitching | ↑fMEPP |
| MFS1 | + | PE | 1: 4800 | + | + |
| MFS2 | + | IVIg | 1: 51200 | + | + |
| GBS1 | + | IVIg | 1: 12800 | + | + |
| GBS2 | + | IVIg | 1: 102400 | + | + |
MFS = Miller Fisher syndrome; GBS = Guillain–Barré syndrome; Ophth = ophthalmoplegia; IgG = immunoglobulin G; ↑fMEPP = increased frequency of miniature end‐plate potentials (>1.8/s); serum dilutions ≥1: 100 (titre anti‐GQ1b IgG) or 1: 2 (twitching, fMEPP); + = present. PE = plasma exchange; IVIg = intravenous immunoglobulins.
Effect of IVIg on twitching and percentage of C3c‐positive end‐plates in mouse diaphragm twitch bioassay induced by serum of four patients with MFS or GBS
| Serum+BSA; NHS+BSA* | Serum+IVIg; NHS+BSA† | Serum+BSA; NHS+IVIg‡ | ||||
| Patient | Twitching | C3c+ | Twitching | C3c+ | Twitching | C3c+ |
| MFS1 | + | 75% | – | 5% | + | 65% |
| MFS2 | + | 60% | – | 10% | + | 45% |
| GBS1 | + | 45% | – | 0% | + | 35% |
| GBS2 | + | 55% | – | 0% | + | 40% |
| Mean (SEM) | 59% (6%) | 4% (2%)§ | 46% (7%) | |||
| Serum+BSA; NHS+BSA* | Serum+IVIg; NHS+BSA† | Serum+BSA; NHS+IVIg‡ | ||||
| Patient | Twitching | C3c+ | Twitching | C3c+ | Twitching | C3c+ |
| MFS1 | + | 75% | – | 5% | + | 65% |
| MFS2 | + | 60% | – | 10% | + | 45% |
| GBS1 | + | 45% | – | 0% | + | 35% |
| GBS2 | + | 55% | – | 0% | + | 40% |
| Mean (SEM) | 59% (6%) | 4% (2%)§ | 46% (7%) | |||
Therapeutic immunoglobulins (25 mg/ml) or bovine serum albumin (25 mg/ml) was simultaneously incubated with patient pre‐treatment serum samples (1:7–1:480) and normal human serum (1:2) as a complement source. Complement activation was determined in 20 end‐plates for each incubation and expressed as the percentage of complement C3c‐positive end‐plates. MFS = Miller Fisher syndrome; GBS = Guillain–Barré syndrome; BSA = bovine serum albumin; NHS = normal human serum; IVIg = intravenous immunoglobulins; + = present; – = absent. Experiments were performed according to *protocol A, †protocol C and ‡protocol F in Fig. 1. §Significantly lower than the group incubated according to protocol A in Fig. 1 (P < 0.05).
Effect of IVIg on twitching and percentage of C3c‐positive end‐plates in mouse diaphragm twitch bioassay induced by serum of four patients with MFS or GBS
| Serum+BSA; NHS+BSA* | Serum+IVIg; NHS+BSA† | Serum+BSA; NHS+IVIg‡ | ||||
| Patient | Twitching | C3c+ | Twitching | C3c+ | Twitching | C3c+ |
| MFS1 | + | 75% | – | 5% | + | 65% |
| MFS2 | + | 60% | – | 10% | + | 45% |
| GBS1 | + | 45% | – | 0% | + | 35% |
| GBS2 | + | 55% | – | 0% | + | 40% |
| Mean (SEM) | 59% (6%) | 4% (2%)§ | 46% (7%) | |||
| Serum+BSA; NHS+BSA* | Serum+IVIg; NHS+BSA† | Serum+BSA; NHS+IVIg‡ | ||||
| Patient | Twitching | C3c+ | Twitching | C3c+ | Twitching | C3c+ |
| MFS1 | + | 75% | – | 5% | + | 65% |
| MFS2 | + | 60% | – | 10% | + | 45% |
| GBS1 | + | 45% | – | 0% | + | 35% |
| GBS2 | + | 55% | – | 0% | + | 40% |
| Mean (SEM) | 59% (6%) | 4% (2%)§ | 46% (7%) | |||
Therapeutic immunoglobulins (25 mg/ml) or bovine serum albumin (25 mg/ml) was simultaneously incubated with patient pre‐treatment serum samples (1:7–1:480) and normal human serum (1:2) as a complement source. Complement activation was determined in 20 end‐plates for each incubation and expressed as the percentage of complement C3c‐positive end‐plates. MFS = Miller Fisher syndrome; GBS = Guillain–Barré syndrome; BSA = bovine serum albumin; NHS = normal human serum; IVIg = intravenous immunoglobulins; + = present; – = absent. Experiments were performed according to *protocol A, †protocol C and ‡protocol F in Fig. 1. §Significantly lower than the group incubated according to protocol A in Fig. 1 (P < 0.05).
Anti‐GQ1b IgG titres and α‐latrotoxin‐like effects in follow‐up sera of three patients with MFS or GBS
| Patient | Time (weeks) | Titre anti‐GQ1b IgG | Twitching | ↑fMEPP | C3c |
| MFS2 | 0 | 1: 51 200 | + | + | 60% |
| 2 | 1: 6400 | – | n.d. | n.d. | |
| 4 | 1: 1600 | – | – | 5% | |
| GBS1 | 0 | 1: 12 800 | + | + | 45% |
| 2 | 1: 1600 | – | – | n.d. | |
| 4 | 1: 400 | – | – | 0% | |
| GBS2 | 0 | 1: 102 400 | + | + | 55% |
| 2 | 1: 3200 | + | + | n.d. | |
| 26 | – | – | – | 0% |
| Patient | Time (weeks) | Titre anti‐GQ1b IgG | Twitching | ↑fMEPP | C3c |
| MFS2 | 0 | 1: 51 200 | + | + | 60% |
| 2 | 1: 6400 | – | n.d. | n.d. | |
| 4 | 1: 1600 | – | – | 5% | |
| GBS1 | 0 | 1: 12 800 | + | + | 45% |
| 2 | 1: 1600 | – | – | n.d. | |
| 4 | 1: 400 | – | – | 0% | |
| GBS2 | 0 | 1: 102 400 | + | + | 55% |
| 2 | 1: 3200 | + | + | n.d. | |
| 26 | – | – | – | 0% |
MFS = Miller Fisher syndrome; GBS = Guillain–Barré syndrome; Time = number of weeks after onset of treatment with intravenous immunoglobulin (0 = pretreatment); IgG = immunoglobulin G; ↑fMEPP = increased frequency of miniature end‐plate potentials (>1.8/s); C3c = percentage of end‐plates positive for staining with anti‐C3c, tested in at least 20 end‐plates (cut‐off value of 27%, i.e. the 97.5 percentile of 11 controls) (O’Hanlon et al., 2001); + = present; – = absent (for anti‐GQ1b antibody titres <1: 100; n.d. = not determined. In the electrophysiological measurements, all folllow‐up sera were tested in 1: 2 dilution.
Anti‐GQ1b IgG titres and α‐latrotoxin‐like effects in follow‐up sera of three patients with MFS or GBS
| Patient | Time (weeks) | Titre anti‐GQ1b IgG | Twitching | ↑fMEPP | C3c |
| MFS2 | 0 | 1: 51 200 | + | + | 60% |
| 2 | 1: 6400 | – | n.d. | n.d. | |
| 4 | 1: 1600 | – | – | 5% | |
| GBS1 | 0 | 1: 12 800 | + | + | 45% |
| 2 | 1: 1600 | – | – | n.d. | |
| 4 | 1: 400 | – | – | 0% | |
| GBS2 | 0 | 1: 102 400 | + | + | 55% |
| 2 | 1: 3200 | + | + | n.d. | |
| 26 | – | – | – | 0% |
| Patient | Time (weeks) | Titre anti‐GQ1b IgG | Twitching | ↑fMEPP | C3c |
| MFS2 | 0 | 1: 51 200 | + | + | 60% |
| 2 | 1: 6400 | – | n.d. | n.d. | |
| 4 | 1: 1600 | – | – | 5% | |
| GBS1 | 0 | 1: 12 800 | + | + | 45% |
| 2 | 1: 1600 | – | – | n.d. | |
| 4 | 1: 400 | – | – | 0% | |
| GBS2 | 0 | 1: 102 400 | + | + | 55% |
| 2 | 1: 3200 | + | + | n.d. | |
| 26 | – | – | – | 0% |
MFS = Miller Fisher syndrome; GBS = Guillain–Barré syndrome; Time = number of weeks after onset of treatment with intravenous immunoglobulin (0 = pretreatment); IgG = immunoglobulin G; ↑fMEPP = increased frequency of miniature end‐plate potentials (>1.8/s); C3c = percentage of end‐plates positive for staining with anti‐C3c, tested in at least 20 end‐plates (cut‐off value of 27%, i.e. the 97.5 percentile of 11 controls) (O’Hanlon et al., 2001); + = present; – = absent (for anti‐GQ1b antibody titres <1: 100; n.d. = not determined. In the electrophysiological measurements, all folllow‐up sera were tested in 1: 2 dilution.
References
Arakawa Y, Yoshimura M, Kobayashi S, Ichihashi K, Miyao M, Momoi MY, et al. The use of intravenous immunoglobulin in Miller Fisher syndrome.
Bain PG, Motomura M, Newsom‐Davis J, Misbah SA, Chapel HM, Lee ML, et al. Effects of intravenous immunoglobulin on muscle weakness and calcium‐channel autoantibodies in the Lambert‐Eaton myasthenic syndrome.
Basta M, Dalakas MC. High‐dose intravenous immunoglobulin exerts its beneficial effect in patients with dermatomyositis by blocking endomysial deposition of activated complement fragments.
Basta M, Fries LF, Frank MM. High doses of intravenous immunoglobulin do not affect the recognition phase of the classical complement pathway.
Basta M, Illa I, Dalakas MC. Increased in vitro uptake of the complement C3b in the serum of patients with Guillain‐Barre syndrome, myasthenia gravis and dermatomyositis.
Basta M, Van Goor F, Luccioli S, Billings EM, Vortmeyer AO, Baranyi L, et al. F(ab)’(2)‐mediated neutralization of C3a and C5a anaphylatoxins: a novel effector function of immunoglobulins.
Buchwald B, Toyka KV, Zielasek J, Weishaupt A, Schweiger S, Dudel J. Neuromuscular blockade by IgG antibodies from patients with Guillain‐Barre syndrome: a macro‐patch‐clamp study.
Buchwald B, Ahangari R, Weishaupt A, Toyka KV. Intravenous immunoglobulins neutralize blocking antibodies in Guillain‐Barre syndrome.
Bullens RW, O’Hanlon GM, Goodyear CS, Molenaar PC, Conner J, Willison HJ, et al. Anti‐GQ1b antibodies and evoked acetylcholine release at mouse motor endplates.
Bullens RW, O’Hanlon GM, Wagner E, Molenaar PC, Furukawa K, Furukawa K, et al. Complex gangliosides at the neuromuscular junction are membrane receptors for autoantibodies and botulinum neurotoxin but redundant for normal synaptic function.
Chapman PB, Houghton AN. Induction of IgG antibodies against GD3 ganglioside in rabbits by an anti‐idiotypic monoclonal antibody.
Chiba A, Kusunoki S, Obata H, Machinami R, Kanazawa I. Serum anti‐GQ1b IgG antibody is associated with ophthalmoplegia in Miller Fisher syndrome and Guillain‐Barre syndrome: clinical and immunohistochemical studies.
Chiba A, Kusunoki S, Obata H, Machinami R, Kanazawa I. Ganglioside composition of the human cranial nerves, with special reference to pathophysiology of Miller Fisher syndrome.
Dalakas MC. Clinical relevance of IVIg in the modulation of the complement‐mediated tissue damage: implications in dermatomyositis, Guillain‐Barre syndrome and myasthenia gravis. In: Kazatchkine MD, Morel D, editors. Intravenous immunoglobulin research and therapy. London: Parthenon;
Dalakas MC. Intravenous immunoglobulin in the treatment of autoimmune neuromuscular diseases: present status and practical therapeutic guidelines.
Dalakas MC, Illa I, Dambrosia JM, Soueidan SA, Stein DP, Otero C, et al. A controlled trial of high‐dose intravenous immune globulin infusions as treatment for dermatomyositis.
Dalakas MC, Fujii M, Li M, Lutfi B, Kyhos J, McElroy B. High‐dose intravenous immune globulin for stiff‐person syndrome.
Dietrich G, Kazatchkine MD. Normal immunoglobulin G (IgG) for therapeutic use (intravenous Ig) contain antiidiotypic specificities against an immunodominant, disease‐associated, cross‐reactive idiotype of human anti‐thyroglobulin autoantibodies.
Dyck PJ, Litchy WJ, Kratz KM, Suarez GA, Low PA, Pineda AA, et al. A plasma exchange versus immune globulin infusion trial in chronic inflammatory demyelinating polyradiculoneuropathy.
Gajdos P, Chevret S, Clair B, Tranchant C, Chastang C. Clinical trial of plasma exchange and high‐dose intravenous immunoglobulin in myasthenia gravis. Myasthenia Gravis Clinical Study Group.
Goodyear CS, O’Hanlon GM, Plomp JJ, Wagner ER, Morrison I, Veitch J, et al. Monoclonal antibodies raised against Guillain‐Barre syndrome‐associated Campylobacter jejuni lipopolysaccharides react with neuronal gangliosides and paralyze muscle‐nerve preparations.
Hafer‐Macko C, Hsieh ST, Li CY, Ho TW, Sheikh K, Cornblath DR, et al. Acute motor axonal neuropathy: an antibody‐mediated attack on axolemma.
Hafer‐Macko CE, Sheikh KA, Li CY, Ho TW, Cornblath DR, McKhann GM, et al. Immune attack on the Schwann cell surface in acute inflammatory demyelinating polyneuropathy.
Ho TW, Hsieh ST, Nachamkin I, Willison HJ, Sheikh K, Kiehlbauch J, et al. Motor nerve terminal degeneration provides a potential mechanism for rapid recovery in acute motor axonal neuropathy after Campylobacter infection.
Jacobs BC, van Doorn PA, Schmitz PI, Tio‐Gillen AP, Herbrink P, Visser LH, et al. Campylobacter jejuni infections and anti‐GM1 antibodies in Guillain‐Barre syndrome.
Jacobs BC, Bullens RW, O’Hanlon GM, Ang CW, Willison HJ, Plomp JJ. Detection and prevalence of alpha‐latrotoxin‐like effects of serum from patients with Guillain‐Barre syndrome.
Jacobs BC, Bullens RW, O’Hanlon GM, Plomp JJ, Willison HJ. Intravenous immunoglobulin inhibits alpha‐latrotoxin‐like effects of Guillain‐Barre syndrome sera.
Kaveri S, Prasad N, Vassilev T, Hurez V, Pashov A, Lacroix‐Desmazes S, et al. Modulation of autoimmune responses by intravenous immunoglobulin (IVIg).
Kazatchkine MD, Kaveri SV. Immunomodulation of autoimmune and inflammatory diseases with intravenous immune globulin.
Kazatchkine MD, Dietrich G, Hurez V, Ronda N, Bellon B, Rossi F, et al. V region‐mediated selection of autoreactive repertoires by intravenous immunoglobulin (i.v.Ig).
Lopez PH, Irazoqui FJ, Nores GA. Normal human plasma contains antibodies that specifically block neuropathy‐associated human anti‐GM1 IgG‐antibodies.
Malik U, Oleksowicz L, Latov N, Cardo LJ. Intravenous gamma‐globulin inhibits binding of anti‐GM1 to its target antigen.
O’Hanlon GM, Bullens RW, Plomp JJ, Willison HJ. Complex gangliosides as autoantibody targets at the neuromuscular junction in Miller Fisher syndrome: a current perspective.
O’Hanlon GM, Plomp JJ, Chakrabarti M, Morrison I, Wagner ER, Goodyear CS, et al. Anti‐GQ1b ganglioside antibodies mediate complement‐dependent destruction of the motor nerve terminal.
Plasma Exchange/Sandoglobulin Guillain‐Barre Syndrome Trial Group. Randomised trial of plasma exchange, intravenous immunoglobulin, and combined treatments in Guillain‐Barre syndrome.
Plomp JJ, Molenaar PC, O’Hanlon GM, Jacobs BC, Veitch J, Daha MR, et al. Miller Fisher anti‐GQ1b antibodies: alpha‐latrotoxin‐like effects on motor end plates.
Roberts M, Willison H, Vincent A, Newsom‐Davis J. Serum factor in Miller‐Fisher variant of Guillain‐Barre syndrome and neurotransmitter release.
Sekul EA, Cupler EJ, Dalakas MC. Aseptic meningitis associated with high‐dose intravenous immunoglobulin therapy: frequency and risk factors.
Shohet JM, Pemberton P, Carroll MC. Identification of a major binding site for complement C3 on the IgG1 heavy chain.
Silbert PL, Knezevic WV, Bridge DT. Cerebral infarction complicating intravenous immunoglobulin therapy for polyneuritis cranialis.
Spaans F, Vredeveld JW, Morre HH, Jacobs BC, De Baets MH. Dysfunction at the motor end‐plate and axon membrane in Guillain‐Barre syndrome: a single‐fiber EMG study.
Tankersley DL, Preston MS, Finlayson JS. Immunoglobulin G dimer: an idiotype‐anti‐idiotype complex.
Teeling JL, Jansen‐Hendriks T, Kuijpers TW, de Haas M, van de Winkel JG, Hack CE, et al. Therapeutic efficacy of intravenous immunoglobulin preparations depends on the immunoglobulin G dimers: studies in experimental immune thrombocytopenia.
Thomas FP, Lee AM, Romas SN, Latov N. Monoclonal IgMs with anti‐Gal(beta
Turner B, Wills AJ. Cerebral infarction complicating intravenous immunoglobulin therapy in a patient with Miller Fisher syndrome.
Uncini A, Lugaresi A. Fisher syndrome with tetraparesis and antibody to GQ1b: evidence for motor nerve terminal block.
Van den Berg LH, Kerkhoff H, Oey PL, Franssen H, Mollee I, Vermeulen M, et al. Treatment of multifocal motor neuropathy with high dose intravenous immunoglobulins: a double blind, placebo controlled study.
van der Meche FG, Schmitz PI. A randomized trial comparing intravenous immune globulin and plasma exchange in Guillain‐Barre syndrome. Dutch Guillain‐Barre Study Group.
Van Doorn PA, Vermeulen M, Brand A, Mulder PG, Busch HF. Intravenous immunoglobulin treatment in patients with chronic inflammatory demyelinating polyneuropathy. Clinical and laboratory characteristics associated with improvement.
Wada J, Shintani N, Kikutani K, Nakae T, Yamauchi T, Takechi K. Intravenous immunoglobulin prevents experimental autoimmune myositis in SJL mice by reducing anti‐myosin antibody and by blocking complement deposition.
Wakamoto H, Ohta M, Nakano N, Tagawa M, Shiraishi T. Intravenous immunoglobulin for cranial polyneuropathy associated with Campylobacter jejuni infection.
Willison HJ, Veitch J, Paterson G, Kennedy PG. Miller Fisher syndrome is associated with serum antibodies to GQ1b ganglioside.
Willison HJ, O’Hanlon GM, Paterson G, Veitch J, Wilson G, Roberts M, et al. A somatically mutated human antiganglioside IgM antibody that induces experimental neuropathy in mice is encoded by the variable region heavy chain gene, V1‐18.
Willison H, Stoll G, Toyka KV, Berger T, Hartung HP. Autoimmunity and inflammation in the peripheral nervous system.
Wirguin I, Ifergane G, Almog Y, Lieberman D, Bersudsky M, Herishanu YO. Presynaptic neuromuscular transmission block in Guillain‐Barre syndrome associated with anti‐GQ1b antibodies.
Yu Z, Lennon VA. Mechanism of intravenous immune globulin therapy in antibody‐mediated autoimmune diseases.
Yuki N, Miyagi F. Possible mechanism of intravenous immunoglobulin treatment on anti‐GM1 antibody‐mediated neuropathies.
Yuki N, Sato S, Tsuji S, Ohsawa T, Miyatake T. Frequent presence of anti‐GQ1b antibody in Fisher’s syndrome.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}