
Abstract
This study investigates aspects of the general assumption that, in bacteria, genetic variation in functionally-constrained genomic regions accumulates at a lower rate than in regions of hypermutability such as DNA repeat loci. We compared whole genome polymorphism (using high-throughput amplified fragment length polymorphism [ht-AFLP]) as well as short sequence repeat length variation (using multi-locus variable number of tandem repeat analysis [MLVA]) for 994 Staphylococcus aureus strains isolated from both healthy carriers and invasive infections. MLVA and ht-AFLP minimum spanning trees (MSTs) were similar in their identification of totally different types of genetic variants. This suggests that, despite the enhanced inherent variability of repeats, clusters of strains remain traceable. Finally, no specific molecular marker of epidemicity or virulence was identified in this large strain collection by the MLVA approach. We demonstrate that there is a difference in the rates of cross-genome mutation versus regional repeat variability in the clonal bacterial pathogen S. aureus. Despite these dynamic differences, a conservation of type assignments as based upon these two inherently different typing techniques was observed.
Similar content being viewed by others


Introduction
Staphylococcus aureus is a relatively clonal microorganism that lives in close contact with human beings [1–3]. Although multiple human body sites can be colonized, the anterior nares are the most frequent carriage site for S. aureus, and nasal carriage appears to play a key role in the epidemiology and pathogenesis of infection [3]. It is assumed that S. aureus’ conserved core genome composition provides the organism with the potential to interact intimately with its human host, whereas the accessory genome, including “core-variable genes”, harbors the specific colonization and virulence genes rendering it infectious [4, 5]. Interestingly, a recent study using microarrays could not identify specific genes, alone or in combination, that were associated with invasive disease. It was suggested that S. aureus gene combinations necessary for invasive disease may also be necessary for nasal colonization and that the ability to cause invasive disease is mostly dependent on host factors [6].
A variety of molecular strategies has been used to distinguish and catalogue core genome versus core-variable and mobile elements in S. aureus [6]. Clonality has been defined on the basis of, among others, multi-locus sequence typing (MLST) and high-throughput amplified fragment length polymorphism (ht-AFLP) studies, whereas core-variable and mobile elements have been identified by, for instance, DNA array hybridization studies [2, 6, 7].
Large scale data obtained by different genome screening methods, focussing on core genomic versus hypermutable regions, have rarely been linked in order to compare molecular type assignments. We provide here an example of such an analysis. We have compared whole genome polymorphism (as defined by ht-AFLP) and short sequence repeat length variation (using multi-locus variable number of tandem repeat analysis [MLVA]) for 994 S. aureus strains isolated from both healthy carriers and invasive infections. Ht-AFLP is a whole genome typing method that scans for polymorphism in restriction sites and the nucleotides bordering these sites. As such it documents nucleotide sequence variation, insertions, and deletions across entire genomes. Recently, MLVA has been introduced as a typing method for a large number of bacterial pathogens [8–10]. In MLVA, the variability in the numbers of short tandem repeated sequences is utilized to create DNA fingerprints for epidemiological studies [9]. We visualize here the genetic variability of S. aureus based on ht-AFLP versus MLVA and define overlaps in the inter-strain relatedness.
Materials and methods
Bacterial isolates and DNA isolation
Carriage and clinical isolates of methicillin-susceptible S. aureus (n = 994) have been described before [7]. Strains analysed in our study included carriage (n = 805), bacteremic (n = 132), skin- and soft-tissue infection (n = 17), and impetigo-derived isolates (n = 40) from children and elderly individuals. These strains were isolated at different time points (between 1997 and 2002) from persons living in the greater Rotterdam area in The Netherlands. DNA was isolated from all isolates using culture on blood agar and automated DNA extraction using the Roche MagnaPure and the Bacterial DNA III isolation kit (Roche, Almere, The Netherlands).
Amplified fragment length polymorphism (AFLP) and multiple-locus variable number of tandem repeat analysis (MLVA)
All strains were genetically typed using a high-throughput AFLP (ht-AFLP) approach as described previously [7]. Optimal enzyme and primer combinations were selected using the predictive software package Recomb (Keygene NV, Wageningen, The Netherlands). Bacterial DNA was digested with the enzymes MboI and Csp6I and the linker oligonucleotide pairs for MboI (5’-CTCGTAGACTGCGTACC-3’ and 5’-GATCGGTACGCAGTCTAC-3’) and for Csp6I (5’-GACGATGAGTCCTGAC-3’ and 5’-TAGTCAGGACTCAT-3’) were ligated. Subsequently, a nonselective preamplification was performed using the MboI (5’-GTAGACTGCGTACCGATC-3’) and Csp6I primers (5’-GACGATGAGTCCTGACTAC-3’). Finally, a 33P-labelled MboI primer containing one selective nucleotide (either +C or +G) and a Csp6I primer containing two selective nucleotides (+TA) were used. Amplified material was analysed using polyacrylamide slabgels and autoradiography. Marker fragments (147 AFLP makers per isolate) were scored and a binary table, scoring marker fragment absence (0) or presence (1), was compiled [7, 11].
In addition, multiple-locus variable number of tandem repeat analysis (MLVA) was performed for all isolates [12, 13]. In short, repetitive DNA from the V8 serine protease (sspA), protein A (spa), Ser-Asp-rich fibrinogen-binding protein (sdrCDE), clumping factor B (clfB), clumping factor A (clfA), fibronectin-binding protein (fnBP), collagen adhesion A (cna), methicillin-resistant surface protein (pls), and cell wall surface-anchored protein (sas) genes were amplified in a single multiplex PCR, also including the nonrepeat containing methicillin resistance (mecA) gene. Amplified material obtained by PCR on purified genomic DNA (1 ng) was subjected to capillary electrophoresis using the Agilent 2100 BioAnalyzer (Agilent, Palo Alto, CA, USA) generating 90 second tracing files containing fluorescence values for amplicons ranging from 50 base pairs to 11 kbp. The BioAnalyzer reagent kit contains upper and lower molecular weight markers used to normalize all profiles.
MLST
Multi-locus sequence typing (MLST) was carried out for a selection of 53 (5.3%) of the 994 S. aureus strains using DNA arrays [14]. These strains were equally distributed across a AFLP dendrogram by selecting approximately one out of 20 strains going from top to bottom through the AFLP dendrogram [7].
Data analysis
For the comparative analyses of both the AFLP and the MLVA data sets, minimum spanning trees (MSTs) were calculated using the Bionumerics software package (Applied-Maths, Sint-Martens-Latem, Belgium). The binary AFLP data were clustered using a categorical coefficient. Complexes were created if the maximum neighbor distance was two changes (except for Fig. 1a which had 16 changes). The MLVA profiles obtained by capillary electrophoresis were normalized based on the lower and upper band present in all samples, and band positions were automatically assigned by the Bionumerics software and manually adjusted where needed. Band based Dice clustering was performed using a 0.5% band position tolerance and 1% optimization. The distance matrix that was thus obtained was used to construct a MST using a 6% similarity bin size to designate MLVA profiles as being identical. For the construction of MLVA complexes a maximum neighbor distance of one change was used.

Minimum spanning trees based on: (a) AFLP data (n = 994) where complexes were created if the maximum neighbour distance was 16 changes, (b) AFLP data (n = 994) where complexes were created if the maximum neighbour distance was two changes, and (c) MLVA data (n = 994) where complexes were created using a cut-off value of 94%
Results and discussion
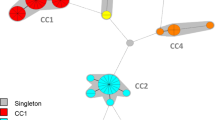
Figure 1 shows the type assignments for all of the strains based on separate analyses of the ht-AFLP and MLVA datasets. The ht-AFLP data of the 994 S. aureus isolates are visualized using two minimum spanning trees (MSTs) with different cut-off values (Fig. 1a and b with a maximum neighbor distance of 16 and two, respectively). It should be emphasized that the (dotted) lines between the clusters do not indicate directional evolutionary relationship between these different clusters. As described earlier, the genuine population structure of S. aureus can be subdivided in three major AFLP clusters (denoted 1, 2, and 3) and two minor AFLP (4 and 5) clusters (Fig. 1a) [7, 15]. In addition, two very small outlier clusters are visible (brown complex [n = 3] and dark blue complex [n = 2]). The AFLP MST in Fig. 1a essentially confirms the clonal structure of S. aureus, which is in contrast with those of other pathogenic species such as Streptococcus pneumoniae [11, 16].
Figure 1b shows that the three major and two minor AFLP clusters could be subdivided in several sub-clusters by applying more stringent segregation parameters during MST construction. The largest cluster (blue) is a genetically heterogeneous cluster in contrast with the two other major clusters (red [2] and green [3]). This is in agreement with the previously reported data on the natural population structure of S. aureus [7]. Major AFLP clusters 2 and 3 are genetically homogeneous lineages and correspond with MLST clonal complexes 30 and 45, respectively. In the analysis where we applied a maximum neighbor distance of two changes for the formation of AFLP complexes or clusters (Figs. 1b, 2a, and 3a), 22 different sub-clusters with more than five S. aureus isolates were identified, and 17 of these sub-clusters or complexes carried more than ten isolates. Several of these clusters coincided with MLST sequence types (ST) (Fig. 2a). Earlier studies showed that AFLP clustering matches with the major clonal complexes as defined by MLST [7, 15].

a Minimum spanning trees based on the AFLP data (n = 994) where MLST data of 53 S. aureus isolates are included in the figure. Each complex in this figure was assigned a different colour. b Minimum spanning trees based on the MLVA data (n = 994) where the colours, which are used in this figure, are based on the different S. aureus AFLP complexes which are shown in Fig. 2a

Minimum spanning trees based on: (a) AFLP data (n = 994) and (b) MLVA data (n = 994). The colours which are used in this figure are based on the source (carriage or invasive) of the S. aureus isolates. Light green are carriage isolates (n = 805); red are “blood isolates” (n = 149) including a few skin- and soft-tissue infection isolates (n = 17); and blue are impetigo-isolates (n = 40)
When the same set of strains was analysed by MLVA (Fig. 1c) the layout of the MST clearly revealed enhanced variability, such that a large number of minor genetic variants are visualized when a cut-off similarity value of 94% was used to identify types. However, the majority of the clonal complexes defined in Fig. 1b co-segregate again on the basis of MLVA. For example, the red, green, and yellow complexes (clusters 2, 3, and 4, respectively) that are identified as closely related by AFLP also remain distinct groups after MLVA-based MST construction. In short, despite the apparently increased complexity of the MLVA MST, there is still similarity with the topology of the AFLP MST. Obviously, deviations can be observed as well.
Finally, Fig. 3 displays the association between the AFLP and MLVA MSTs and the clinical origin of the strains. The conclusion is that both invasive and colonizing isolates are present in all S. aureus clusters, whether these are defined by AFLP (Fig. 3a) or MLVA (Fig. 3b). In cases of the impetigo isolates, a certain degree of clonal expansion could be hypothesized on the basis of the presence of several strains in a single AFLP cluster. Furthermore, some of the AFLP clusters (Fig. 3a) show a slight overrepresentation of bacteremia isolates compared to the average number of such isolates in the other clusters, which corroborates earlier data [7]. However, it should be noted that studies using MLST data could not identify hypervirulent clones of S. aureus [2, 17]. In addition, a recent study using microarrays was unable to identify any association between lineage or gene and invasive isolates [6].
It is thought that the generation of genetic variation in hypermutable loci such as short sequence repeats proceeds with enhanced speed, even in clonal species such as S. aureus [18]. We show here that there indeed is a difference in the rates or speed of whole genome versus repeat variation in S. aureus with repeat-derived type assignments showing enhanced diversification (see Fig. 2a versus b). However, a clear topological overlap in the MSTs calculated on the basis of complex repeat patterns versus those calculated on the basis of whole genome polymorphism can still be observed. This suggests that even hypervariation of repeat loci remains within the framework set out by overall genome variation. This is in agreement with earlier studies showing that typing techniques using highly variable genes (e.g. adhesion genes) are at least as informative for phylogenetic reconstruction as the more slowly evolving housekeeping genes (e.g. MLST) [19–23].
In conclusion, we demonstrate here that for a clonal bacterial pathogen such as S. aureus, analysis of DNA targets with different inherent degrees of genetic variability results in overlapping molecular type assignments. This suggests that despite the enhanced variability of repeats, clusters of strains remain traceable. In other words, measuring repeat polymorphisms in clonal microorganisms provides a solid basis for genetic type assignment useful for application in epidemiological tracing.
References
Enright MC, Day NP, Davies CE, Peacock SJ, Spratt BG (2000) Multilocus sequence typing for characterization of methicillin-resistant and methicillin-susceptible clones of Staphylococcus aureus. J Clin Microbiol 38(3):1008–1015
Feil EJ, Cooper JE, Grundmann H, Robinson DA, Enright MC, Berendt T et al (2003) How clonal is Staphylococcus aureus? J Bacteriol 185(11):3307–3316. doi:10.1128/JB.185.11.3307-3316.2003
Wertheim HF, Melles DC, Vos MC, van Leeuwen W, van Belkum A, Verbrugh HA et al (2005) The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect Dis 5(12):751–762. doi:10.1016/S1473-3099(05)70295-4
Lina G, Piemont Y, Godail-Gamot F, Bes M, Peter MO, Gauduchon V et al (1999) Involvement of Panton-Valentine leukocidin-producing Staphylococcus aureus in primary skin infections and pneumonia. Clin Infect Dis 29(5):1128–1132. doi:10.1086/313461
Peacock SJ, Moore CE, Justice A, Kantzanou M, Story L, Mackie K et al (2002) Virulent combinations of adhesin and toxin genes in natural populations of Staphylococcus aureus. Infect Immun 70(9):4987–4996. doi:10.1128/IAI.70.9.4987-4996.2002
Lindsay JA, Moore CE, Day NP, Peacock SJ, Witney AA, Stabler RA et al (2006) Microarrays reveal that each of the ten dominant lineages of Staphylococcus aureus has a unique combination of surface-associated and regulatory genes. J Bacteriol 188(2):669–676. doi:10.1128/JB.188.2.669-676.2006
Melles DC, Gorkink RF, Boelens HA, Snijders SV, Peeters JK, Moorhouse MJ et al (2004) Natural population dynamics and expansion of pathogenic clones of Staphylococcus aureus. J Clin Invest 114(12):1732–1740
Schouls LM, van der Ende A, Damen M, van de Pol I (2006) Multiple-locus variable-number tandem repeat analysis of Neisseria meningitidis yields groupings similar to those obtained by multilocus sequence typing. J Clin Microbiol 44(4):1509–1518. doi:10.1128/JCM.44.4.1509-1518.2006
Tenover FC, Vaughn RR, McDougal LK, Fosheim GE, McGowan JE (2007) Multiple-locus variable-number tandem-repeat assay analysis of methicillin-resistant Staphylococcus aureus strains. J Clin Microbiol 45(7):2215–2219. doi:10.1128/JCM.02451-06
van Belkum A (2007) Tracing isolates of bacterial species by multilocus variable number of tandem repeat analysis (MLVA). FEMS Immunol Med Microbiol 49(1):22–27. doi:10.1111/j.1574-695X.2006.00173.x
Melles DC, Bogaert D, Gorkink RF, Peeters JK, Moorhouse MJ, Ott A et al (2007) Nasopharyngeal co-colonization with Staphylococcus aureus and Streptococcus pneumoniae in children is bacterial genotype independent. Microbiology 153(Pt 3):686–692. doi:10.1099/mic.0.2006/002279-0
Francois P, Huyghe A, Charbonnier Y, Bento M, Herzig S, Topolski I et al (2005) Use of an automated multiple-locus, variable-number tandem repeat-based method for rapid and high-throughput genotyping of Staphylococcus aureus isolates. J Clin Microbiol 43(7):3346–3355. doi:10.1128/JCM.43.7.3346-3355.2005
Koessler T, Francois P, Charbonnier Y, Huyghe A, Bento M, Dharan S et al (2006) Use of oligoarrays for characterization of community-onset methicillin-resistant Staphylococcus aureus. J Clin Microbiol 44(3):1040–1048. doi:10.1128/JCM.44.3.1040-1048.2006
van Leeuwen WB, Jay C, Snijders S, Durin N, Lacroix B, Verbrugh HA et al (2003) Multilocus sequence typing of Staphylococcus aureus with DNA array technology. J Clin Microbiol 41(7):3323–3326. doi:10.1128/JCM.41.7.3323-3326.2003
Melles DC, van Leeuwen WB, Snijders SV, Horst-Kreft D, Peeters JK, Verbrugh HA et al (2007) Comparison of multilocus sequence typing (MLST), pulsed-field gel electrophoresis (PFGE), and amplified fragment length polymorphism (AFLP) for genetic typing of Staphylococcus aureus. J Microbiol Methods 69(2):371–375. doi:10.1016/j.mimet.2007.01.013
Feil EJ, Smith JM, Enright MC, Spratt BG (2000) Estimating recombinational parameters in Streptococcus pneumoniae from multilocus sequence typing data. Genetics 154(4):1439–1450
Turner KM, Feil EJ (2007) The secret life of the multilocus sequence type. Int J Antimicrob Agents 29(2):129–135. doi:10.1016/j.ijantimicag.2006.11.002
van Belkum A, Scherer S, van Alphen L, Verbrugh H (1998) Short-sequence DNA repeats in prokaryotic genomes. Microbiol Mol Biol Rev 62(2):275–293
Cooper JE, Feil EJ (2006) The phylogeny of Staphylococcus aureus—which genes make the best intra-species markers? Microbiology 152(Pt 5):1297–1305. doi:10.1099/mic.0.28620-0
Gomes AR, Vinga S, Zavolan M, de Lencastre H (2005) Analysis of the genetic variability of virulence-related loci in epidemic clones of methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 49(1):366– 379. doi:10.1128/AAC.49.1.366-379.2005
Kuhn G, Francioli P, Blanc DS (2006) Evidence for clonal evolution among highly polymorphic genes in methicillin-resistant Staphylococcus aureus. J Bacteriol 188(1):169–178. doi:10.1128/JB.188.1.169-178.2006
Robinson DA, Monk AB, Cooper JE, Feil EJ, Enright MC (2005) Evolutionary genetics of the accessory gene regulator (agr) locus in Staphylococcus aureus. J Bacteriol 187(24):8312–8321. doi:10.1128/JB.187.24.8312-8321.2005
Mellmann A, Weniger T, Berssenbrugge C, Rothganger J, Sammeth M, Stoye J et al (2007) Based upon repeat pattern (BURP): an algorithm to characterize the long-term evolution of Staphylococcus aureus populations based on spa polymorphisms. BMC Microbiol 7:98. doi:10.1186/1471-2180-7-98
Acknowledgements
Members of the Department of Medical Microbiology and Infectious Diseases (Erasmus MC, Rotterdam, The Netherlands) are thanked for support provided during various stages of the AFLP project (BTS 00145). This work was also supported by grants from the Swiss National Science Foundation #PP00B–103002/1 and # 3100A0–112370/1 (JS), # 404940–106296/1 and # 3100A0–116075/1 (PF). AFLP is a registered trademark of Keygene N.V. and the AFLP technology is covered by patents (US006045994A, EP0534858B1) and patent applications owned by Keygene N.V. The REcomb software is covered by patent applications (WO 00/44937) of Keygene N.V.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Melles, D.C., Schouls, L., François, P. et al. High-throughput typing of Staphylococcus aureus by amplified fragment length polymorphism (AFLP) or multi-locus variable number of tandem repeat analysis (MLVA) reveals consistent strain relatedness. Eur J Clin Microbiol Infect Dis 28, 39–45 (2009). https://doi.org/10.1007/s10096-008-0585-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10096-008-0585-4