

Abstract
Background
Pediatric drug development is hampered by practical, ethical, and scientific challenges. Microdosing is a promising new method to obtain pharmacokinetic data in children with minimal burden and minimal risk. The use of a labeled oral microdose offers the added benefit to study intestinal and hepatic drug disposition in children already receiving an intravenous therapeutic drug dose for clinical reasons.
Objective
The objective of this study was to present pilot data of an oral [14C]paracetamol [acetaminophen (AAP)] microdosing study as proof of concept to study developmental pharmacokinetics in children.
Methods
In an open-label microdose pharmacokinetic pilot study, infants (0–6 years of age) received a single oral [14C]AAP microdose (3.3 ng/kg, 60 Bq/kg) in addition to intravenous therapeutic doses of AAP (15 mg/kg intravenous every 6 h). Blood samples were taken from an indwelling catheter. AAP blood concentrations were measured by liquid chromatography–tandem mass spectrometry (LC-MS/MS) and [14C]AAP and metabolites ([14C]AAP-Glu and [14C]AAP-4Sul) were measured by accelerator mass spectrometry.
Results
Ten infants (aged 0.1–83.1 months) were included; one was excluded as he vomited shortly after administration. In nine patients, [14C]AAP and metabolites in blood samples were detectable at expected concentrations: median (range) maximum concentration (C max) [14C]AAP 1.68 (0.75–4.76) ng/L, [14C]AAP-Glu 0.88 (0.34–1.55) ng/L, and [14C]AAP-4Sul 0.81 (0.29–2.10) ng/L. Dose-normalized oral [14C]AAP C max approached median intravenous average concentrations (C av): 8.41 mg/L (3.75–23.78 mg/L) and 8.87 mg/L (3.45–12.9 mg/L), respectively.
Conclusions
We demonstrate the feasibility of using a [14C]labeled microdose to study AAP pharmacokinetics, including metabolite disposition, in young children.
Electronic supplementary material
The online version of this article (doi:10.1007/s40262-014-0176-8) contains supplementary material, which is available to authorized users.
Key Points
| A [14C]labeled microdosing study is feasible in young children. |
| [14C]Labeled microdosing may be used to study developmental pharmacokinetics, including oral bioavailability. |
Introduction
Up to 70 % of drugs prescribed to children are unlicensed or off-label, which brings risk of drug toxicity or therapeutic failure [1, 2]. However, pediatric drug studies face important ethical, practical, and scientific challenges [3]. A major challenge—against the background of developmental changes in drug absorption, distribution, metabolism, and excretion—is appropriate dose selection [4]. Simple size- or weight-based extrapolations from adult to pediatric doses do not suffice, particularly in neonates and infants. Current strategies include simulations using population pharmacokinetic (popPK) and physiologically based pharmacokinetic (PBPK) models [5–9]. The usefulness of these models may be limited, as relatively little pediatric pharmacokinetic and physiological data are available to validate them. Hence, new data are needed to support these models, as well as alternative methods to collect pharmacokinetic data without the inherent risks of toxicity when a therapeutic dose is given for the first time in a specific age group.
Microdosing is an interesting alternative. The European Medicines Agency (EMA) and the US Food and Drug Administration (FDA) define a microdose as one-hundredth of the No Observed Adverse Effect Level (NOAEL) or predicted pharmacologic dose based on animal data or as 100 μg of the new drug, whichever dose is lower [10, 11]. Dose linearity between the microdose and therapeutic dose is a prerequisite to extrapolate pharmacokinetic data to dosing guidelines [12]. The extremely low dose concentrations call for highly sensitive measurements. Accelerator mass spectrometry (AMS) can measure low attomolar to zeptomolar isotope ratio ranges, to quantify [14C]labeled drug or metabolite concentrations in urine or plasma samples, even after at least five half-lives following a microdose [13]. [14C] Labeling of a microdose is associated with very low radiation exposure, i.e., less than 10 μSv in adults, when compared with the yearly background exposure of 2.5 mSv/year in The Netherlands [14]. Hence, microdosing is safe to use to study pharmacokinetics in children. In clinical care, [14C]urea has been safely used to test for Helicobacter pylori infection [15]. A [14C]ursodiol microdose study in preterm infants was briefly described in an excellent review on the potential for use of AMS in children [16]. Administering a labeled oral microdose to children already receiving a therapeutic drug dose for clinical reasons intravenously offers the added possibility to study oral bioavailability [12, 17]. This approach may also serve to delineate developmental changes in the drug-metabolizing enzymes involved in intestinal and hepatic drug metabolism [17, 18].
Paracetamol (acetaminophen; AAP), is much used in children and is an interesting study drug for several reasons. First, its metabolism shifts from primarily sulphation (AAP-sulphate [AAP-Sul]) to glucuronidation (AAP-glucuronide [AAP-Glu]) in the first year of life, as reflected by urinary metabolite kinetics [19, 20]. The relative contribution of intestinal and hepatic drug metabolism has not been studied to date. Second, dose linearity under normal conditions and after probenecid glucuronidation inhibition was shown in adults [18]. We therefore selected oral [14C]AAP to study the developmental changes in AAP glucuronidation and sulphation in children already receiving the non-labeled or ‘cold’ drug intravenously for analgesia [14]. This paper presents our first pilot data as proof of concept of this promising, safe method to study developmental pharmacokinetics in children.
Methods
Study Design
This proof-of-concept study is part of a larger phenotyping study in 60 children delineating developmental changes in AAP to metabolite clearance (EudraCT 2011-005497-28). The Dutch Central Committee on Research Involving Human Subjects (The Hague, The Netherlands) approved the larger study. Parents or legal guardians gave informed consent. Radiation exposure was explained during the informed consent procedure in relation to background exposure and exposure from medical imaging. The Dutch Nuclear Research and Service Group estimated individual exposures at approximately 1 μSv for a 0- to 2-year-old child, well below the minimal risk category 1 (100 μSv) of the International Commission of Radiological Protection and the yearly background exposure (2.5 mSv). Category 1 risk studies are considered minimal risk and are allowed when they provide new scientific knowledge [11].
Subjects
Patients admitted to the Intensive Care department of the Erasmus MC-Sophia Children’s Hospital (Rotterdam, The Netherlands) were considered for inclusion. Inclusion criteria were age between 0 and 6 years, gestational age >36 weeks, medical need for intravenous AAP, and an indwelling central venous or arterial line in place. To reduce pharmacokinetic variability due to underlying disease, patients with kidney and liver injury or the use of more than one vasopressor drug were excluded, as well as children intolerant to enteral nutrition.
Study Procedures
A single oral [14C]AAP microdose (3.3 ng/kg, 60 Bq/kg, 0.25 mL/kg) followed by 1 mL of saline (to ensure rinsing of the enteral feeding tube) was administered in addition to the intravenous therapeutic dose of AAP (15 mg/kg intravenously every 6 h) prescribed by the treating physician to provide analgesia. The [14C]AAP oral dose also contained 1.7 µg/kg non-labeled AAP, but this amount was negligible in relation to the therapeutic intravenous dose and thus considered irrelevant for the pharmacokinetic estimations. The microdose was based on a previous adult [14C]AAP microdose study using 100 μg/7.1 kBq/individual and was normalized for weight [21]. Blood samples (0.5 mL) were taken from the indwelling catheter before and at 10–30 min, 1, 2, 4, 6, 12, and 24 h after dosing. Blood samples were centrifuged and plasma was stored at −80 °C until analysis.
Medicinal Products
The AAP formulation (10 mg/mL) was purchased from Fresenius Kabi, Schelle, Belgium. [14C]AAP was purchased from Moravek Biochemicals (Brea, CA, USA).
Radiopharmaceutical Preparation
The formulation for oral administration [14C]AAP was prepared by adding [14C]AAP to an AAP formulation for intravenous use at the good manufacturing practice (GMP) radiopharmaceutical production laboratories of the Department of Radiology and Nuclear Medicine at the VU University Medical Center (Amsterdam, The Netherlands) (GMP license no. NL/H 11/0005) at final concentrations of 13 ng/mL for [14C]AAP and 6.7 µg/mL for non-labeled AAP. The mixed formulation was passed over a Millex®-GV 0.22 µm filter and dispended in 20 mL sterile vials. Radiochemical purity was >99 %; chemical purity >98 %. The radiopharmaceutical was heat sterilized and was shown to be stable for 2 months.
Non-specific binding was tested by running the [14C]AAP formulation followed by 1 mL of saline through enteral feeding tubes. Radioactive recovery measured by liquid scintillation was greater than 95 %.
Paracetamol (Acetaminophen; AAP) Analysis
AAP concentrations were measured in the Hospital Pharmacy laboratory of Erasmus MC with a clinically used enzyme multiplied immunoassay technique (EMIT, Abbott Laboratories, Weesp, the Netherlands) immunoassay method (Abbott Laboratories®) with a lower limit of quantification (LLOQ) of 2.8 mg/L.
[14C]AAP and Metabolite Analysis
Plasma Sample Extraction and Ultra Performance Liquid Chromatography Separation
Using 175 μL 100 % v/v methanol containing 6.6 μg/mL of AAP in 96-well protein precipitation plates, 45 μL of plasma was extracted. The pellet was washed with 100 μL 0.9 % NaCl :100 % methanol (1:4 v/v). Resulting filtrates were evaporated to dryness, and re-dissolved in 30 μL 10 mM ammonium phosphate pH3.4 (Eluent A) of which 25 μL was used for ultra performance liquid chromatography (UPLC) analysis. An AAP solution with a specific radioactivity of 4,100 Bq [14C]AAP/100 μg AAP in blank pooled plasma was used to prepare eight calibrators levels and three quality control (QC) sample levels from 0.4 to 180 mBq/mL, and from 1.7 to 131 mBq/mL, respectively. Calibrators (duplicate), QCs (triplicate), and sample extracts were injected onto a UPLC coupled to a photodiode array (PDA). Chromatographic conditions can be found in Table 1. AAP in 100 % methanol was added to each collected fraction to increase the carbon-12 content to 25 μg.
Table 1.
Ultra performance liquid chromatography conditions for paracetamol (acetaminophen)
| Eluent A | 10 mM ammonium phosphate pH 3.4 |
| Eluent B | 100 % v/v methanol |
| UPLC column (Waters Acquity) | BEH C18 1.7 μm 2.1 × 100 mm column |
| Flowrate | 0.3 mL/min |
| Column temperature | 30 °C |
| Pressure | 700 bar |
| Chromatography conditions | 0–1 min 100 % A and 0 % B |
| 1–10 min linear gradient from 100 % A and 0 % B to 95 % A and 5 % B | |
| 10–12 min 95 % A and 5 % B | |
| 12–15 min linear gradient from 95 % A and 5 % B to 0 % A and 100 % B | |
| 15–20 min 0 % A and 100 % B | |
| 20–20.10 min linear gradient from 0 % A and 100 % B to 100 % A and 0 % B | |
| 20.10–20.50 min 100 %A and 0 % B | |
| 20.50–28 min 100 % A and 0 % B at a flowrate of 0.4 mL/min | |
| 28–29 min 100 % A and 0 % B | |
| Collected fractions | [14C]AAP-Glu (3.8–5.3 min) |
| [14C]AAP-4Sul (6.1–7.9 min) | |
| [14C]AAP (8.1–9.8 min) |
UPLC ultra performance liquid chromatography
Accelerator Mass Spectrometry Analysis
[14C]AAP and metabolites were determined as described recently [22]. A novel AMS sample introduction method was used in this study to allow the routine analysis of biomedical samples. The method consists of an automated carbon dioxide (CO2) combustion device online coupled to an AMS. Briefly, dried fractionated samples are placed in a tin-foil cup and subsequently combusted using an elemental analyzer (EA) (Vario Micro, Elementar, Germany). The resulting CO2 was captured on a zeolyte trap at the interface, connecting the EA to the AMS. CO2 was released by heating of the trap and transferred to a vacuum syringe using helium. The resulting 6 % v/v gas mixture of CO2 with helium was infused at a pressure of 1 bar at 60 μL/min into the titanium target in the SO110 ion source of a 1 MV Tandetron AMS (High Voltage Engineering Europe B.V., Amersfoort, The Netherlands) [23]. Within the source, CO2 is converted into negative carbon ions. The validation for the liquid chromatography (LC) + AMS analysis was based on the recommendation of the European Bioanalytical Forum. Three QC concentration levels were included: QC High 145 mBq/mL, QC Medium 14.5 mBq/mL, and QC Low 2.4 mBq/mL. The accuracy of QC High, Medium, and Low analysis corresponded to 104, 103, and 90 %, respectively. The precisions, defined by a coefficient of variation, were 9.1, 6.7, and 6.9 %, respectively. All values are well within the requirements for LC + AMS analysis [24]. The LLOQ of the method was 0.58 mBq/mL.
Data Analysis
Data were summarized as median (range), unless noted otherwise. The plasma [14C]AAP and metabolite concentrations were calculated by converting measured Bq/L to ng/L based on the dose given (3.3 ng/kg contained 60 Bq/kg) and for the metabolites corrected for molecular weight ([14C]AAP 151 g/mol, [14C]AAP-Glu 237 g/mol, and [14C]AAP-4Sul 231 g/mol) To compare the disposition of oral [14C]AAP microdose with the intravenous therapeutic doses, the microdose concentrations were dose normalized to 15 mg/kg by multiplying with 5 × 10E6.
Results
Patients
Between 13 January and 31 May 2014, 32 patients were eligible. Nine patients were excluded for logistical reasons and 13 for refusal of informed consent. Parents’ informed consent was received for ten patients who were subsequently dosed according to the protocol. One patient vomited within 15 min post-dose and was excluded from pharmacokinetic analysis. Table 2 provides a summary of patient characteristics.
Table 2.
Patient characteristics
| Patient | Post-natal age (months) | Sex | Primary diagnosis | Intervention |
|---|---|---|---|---|
| 1 | 3.6 | Male | Post-necrotizing enterocolitis sigmoïd stenosis | Post-operative partial proximal colon resection |
| 2 | 10.6 | Female | Scaphocephaly | Post-operative craniofacial correction |
| 3 | 1.7 | Female | Congenital cystic adenomatoid malformation of the lung | Post-operative partial lung resection |
| 4 | 0.3 | Male | Congenital diaphragmatic hernia | Post-operative hernia correction |
| 5 | 53.8 | Male | Scaphocephaly | Post-operative craniofacial correction |
| 6 | 28.9 | Male | Germ cell tumor | Respiratory insufficiency due to mediastinal pressure |
| 7a | 83.1 | Male | Increased intracranial pressure | Post-operative craniofacial correction |
| 8 | 0.1 | Male | Congenital cardiac disease | Monitoring respiratory insufficiency |
| 9 | 6.2 | Male | Scaphocephaly | Post-operative craniofacial correction |
| 10 | 5.6 | Male | Duodenal web | Post-operative duodenoduodenostomy and placing gastrostomy tube |
aPatient 7 excluded from results after vomiting within 15 min after microdose intake
Detection of [14C]AAP and Metabolites in Plasma
Time profiles of [14C]AAP, [14C]AAP-Glu, and [14C]AAP-4Sul metabolite plasma concentrations of two representative patients are shown in Fig. 1. These patients were chosen as they represent the youngest and the oldest age group. The Electronic Supplementary Material shows graphs of all patients. The median time to maximum concentration (t max) and maximum concentration (C max) median (range) for [14C]AAP were, respectively, 153 min (10–245 min) and 1.68 ng/L (0.75–4.76 ng/L), for [14C]AAP-Glu were 248 min (161–382 min) and 0.88 ng/L (0.34–1.55 ng/L), and for [14C]AAP-4Sul were 193 min (115–343 min) and 0.81 ng/L (0.29–2.10 ng/L). Sample collections were not complete for all patients as the arterial line was prematurely removed or access to the line was restricted for clinical reasons.
Fig. 1.
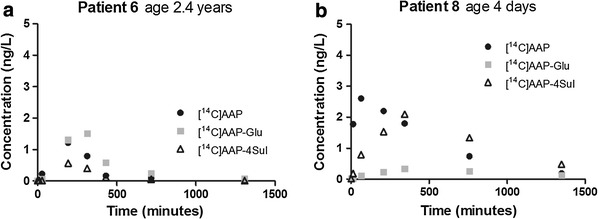
[14C]AAP and metabolite plasma concentrations after an oral [14C]AAP 3.3 ng/kg dose (lower limit of quantification 0.03 ng/L). AAP acetaminophen (paracetamol), Glu glucuronide, Sul sulphate
Dose-Normalized [14C]AAP and AAP Disposition
Semilog plots of the same two patients of dose-normalized 3.3 ng/kg oral [14C]AAP and 15 mg/kg every 6 h intravenous AAP concentrations are shown in Fig. 2. Individual graphs of all patients are displayed in the Electronic Supplementary Material. Dose-normalized median [14C]AAP C max concentrations approached median intravenous average concentrations (C av) [median (range)]: 8.41 mg/L (3.75–23.78 mg/L) and 8.87 mg/L (3.45–12.9 mg/L), respectively.
Fig. 2.
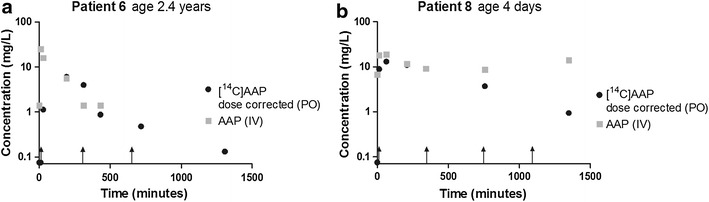
Dose-normalized plasma concentrations after a PO [14C]AAP (3.3 ng/kg) and IV AAP (15 mg/kg/ every 6 h) dose (dose-normalized LLOQ AMS 0.03 mg/L; LLOQ LC-MS/MS 2.8 mg/L). AAP IV administration timepoints are indicated with black arrows. Patient 6: at the latter two timepoints there was insufficient blood collected to analyze AAP concentrations in plasma. AAP acetaminophen (paracetamol), AMS accelerator mass spectrometry, IV intravenous, LC-MS/MS liquid chromatography–tandem mass spectrometry, LLOQ lower limit of quantification, PO oral
Discussion
This proof-of-concept study shows the practical and ethical feasibility of a [14C]microdose study in children to study developmental pharmacokinetics.
The concentrations of [14C]AAP are in the expected range, when compared with dose-normalized concentrations previously reported in neonates and children [25–27]. The average [14C]AAP concentrations and metabolite C max values are also similar to those in a previous adult study: 1.64 versus 1.68 ng/mL for [14C]AAP, 0.88 versus 0.92 ng/mL for [14C]AAP-Glu, and 0.62 versus 0.88 ng/mL for [14C]AAP-4Sul [18]. The apparent lack of an age-related difference in metabolite disposition is easily explained by our small sample size and the even smaller number of neonates in our cohort. This is supported by our observation that in the 4-day-old neonate [14C]AAP-4Sul concentrations are much higher than the [14C]AAP-Glu concentrations, while the opposite is observed in the 2.4-year-old, in line with developmental changes in AAP metabolism. In the follow-up study up to 50 additional patients will be included to study developmental changes in AAP disposition with enough statistical power. Interestingly, the average t max values in our patients are much later than in the adult study: 0.25 versus 2.6 h for [14C]AAP, 0.25 versus 4 h for [14C]AAP-Glu, and 0.5 versus 3.2 h for [14C]AAP-4Sul. A possible explanation for this finding may be slower oral absorption due to maturation and the underlying critical illness or post-operative state in our patients [26, 28].
The major barrier to a [14C]microdosing study in children has been the perceived risk of radiation in the context of a non-therapeutic trial [29, 30]. Still, radiation exposure in this study was extremely low (<1 μSv), much lower than yearly background exposure (2.5 mSv/year in The Netherlands), a continental flight (>4 μSv), exposure from chest X-ray (10 μSv), or computed tomography scans (100 μSv) [7, 14]. To overcome parental ethical barriers to the study, we added a letter from the Dutch collaborative patients’ organization for rare and genetic diseases (VSOP) to the patient information leaflet, explaining the need for pediatric drug research and the minimal risk involved in this study. Surprisingly, from the informed consent conversations it appeared that most parents perceived there was minimal risk involved. Fear for harmful radiation exposure was not the main reason to deny informed consent. Parents of the other children refused informed consent for reasons relating to the burden of additional procedures and/or blood sampling.
Very sensitive LC–tandem mass spectrometry (LC-MS/MS) techniques to measure the pharmacokinetics of an unlabeled microdose may be an alternative to using a radiolabeled microdose in general [31]. Nevertheless, while low drug concentrations can be measured with LC-MS/MS, this analytical method has not reached the very low limits of detection of AMS. Hence, a larger unlabeled dose may be needed, which may increase the risk of a therapeutic or toxic effect [13]. Moreover, the use of an unlabeled microdose would also prohibit the separation of the disposition of an oral and intravenous dose given at the same time. This could be overcome by using stable isotope-labeled probe drugs, which have successfully been used to study oral bioavailability using 15N3-midazolam in adults [32]. This method also has the important disadvantage that a much higher labeled drug dose is needed with similar or even higher risk of therapeutic or toxic effect.
Conclusion
We have shown proof of concept for the practical and ethical feasibility of a [14C]labeled microdose to study pharmacokinetics in young children. This approach offers innovative possibilities to perform phase I first-in-child studies, especially for drugs with a small therapeutic window and high toxicity. In addition, it enables studies in vulnerable populations such as critically ill neonates and studies on developmental pharmacokinetics using probe drugs for specific elimination pathways such as drug-metabolizing enzymes and renal excretion.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
We would like to thank Ko Hagoort for critically reading the manuscript. This work was funded by a Netherlands Organization for Health Research and Development (ZonMw) research grant (113202007). All authors have no potential conflicts of interest to declare.
Contributor Information
Edwin Spaans, Email: e.spaans@erasmusmc.nl.
Saskia N. de Wildt, Phone: +31 10 70 36 889, Email: s.dewildt@erasmusmc.nl
References
- 1.Schirm E, Tobi H, de Vries TW, Choonara I, De Jong-van den Berg LT. Lack of appropriate formulations of medicines for children in the community. Acta Paediatr. 2003;92(12):1486–1489. doi: 10.1111/j.1651-2227.2003.tb00837.x. [DOI] [PubMed] [Google Scholar]
- 2.t Jong GW, van der Linden PD, Bakker EM, van der Lely N, Eland IA, Stricker BH. Unlicensed and off-label drug use in a paediatric ward of a general hospital in the Netherlands. Eur J Clin Pharmacol. 2002;58(4):293–297. doi: 10.1007/s00228-002-0479-9. [DOI] [PubMed] [Google Scholar]
- 3.Hoppu K. Reflection: medicines for children–science alone is not enough. Eur J Clin Pharmacol. 2013;69(Suppl 1):59–63. doi: 10.1007/s00228-013-1487-7. [DOI] [PubMed] [Google Scholar]
- 4.Kearns GL, Abdel-Rahman SM, Alander SW, Blowey DL, Leeder JS, Kauffman RE. Developmental pharmacology–drug disposition, action, and therapy in infants and children. N Engl J Med. 2003;349(12):1157–1167. doi: 10.1056/NEJMra035092. [DOI] [PubMed] [Google Scholar]
- 5.Krekels EH, Danhof M, Tibboel D, Knibbe CA. Ontogeny of hepatic glucuronidation; methods and results. Curr Drug Metab. 2012;13(6):728–743. doi: 10.2174/138920012800840455. [DOI] [PubMed] [Google Scholar]
- 6.De Cock RF, Piana C, Krekels EH, Danhof M, Allegaert K, Knibbe CA. The role of population PK-PD modelling in paediatric clinical research. Eur J Clin Pharmacol. 2011;67(Suppl 1):5–16. doi: 10.1007/s00228-009-0782-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barrett JS, Della Casa Alberighi O, Laer S, Meibohm B. Physiologically based pharmacokinetic (PBPK) modeling in children. Clin Pharmacol Ther. 2012;92(1):40–49. doi: 10.1038/clpt.2012.64. [DOI] [PubMed] [Google Scholar]
- 8.Knibbe CA, Danhof M. Individualized dosing regimens in children based on population PKPD modelling: are we ready for it? Int J Pharm. 2011;415(1–2):9–14. doi: 10.1016/j.ijpharm.2011.02.056. [DOI] [PubMed] [Google Scholar]
- 9.Knibbe CA, Krekels EH, Danhof M. Advances in paediatric pharmacokinetics. Expert Opin Drug Metab Toxicol. 2011;7(1):1–8. doi: 10.1517/17425255.2011.539201. [DOI] [PubMed] [Google Scholar]
- 10.Food and Drug Administration US Department of Health and Human Services. Guidance for Industry Investigators and Reviewers. Exploratory IND Studies. January 2006. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm078933.pdf. Accessed 25 Aug 2014.
- 11.European Medicines Agency. ICH topic M3 (R2). Non-clinical safety studies for the conduct of human clinical trials and marketing authorization for pharmaceuticals. London: European Medicines Agency; Jul 2008.
- 12.Lappin G, Kuhnz W, Jochemsen R, Kneer J, Chaudhary A, Oosterhuis B, et al. Use of microdosing to predict pharmacokinetics at the therapeutic dose: experience with 5 drugs. Clin Pharmacol Ther. 2006;80(3):203–215. doi: 10.1016/j.clpt.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 13.Salehpour M, Possnert G, Bryhni H. Subattomole sensitivity in biological accelerator mass spectrometry. Anal Chem. 2008;80(10):3515–3521. doi: 10.1021/ac800174j. [DOI] [PubMed] [Google Scholar]
- 14.Ceelie I, de Wildt SN, van Dijk M, van den Berg MM, van den Bosch GE, Duivenvoorden HJ, et al. Effect of intravenous paracetamol on postoperative morphine requirements in neonates and infants undergoing major noncardiac surgery: a randomized controlled trial. JAMA. 2013;309(2):149–154. doi: 10.1001/jama.2012.148050. [DOI] [PubMed] [Google Scholar]
- 15.Pathak CM, Kaur B, Khanduja KL. 14C-urea breath test is safe for pediatric patients. Nucl Med Commun. 2010;31(9):830–835. doi: 10.1097/MNM.0b013e32833c3647. [DOI] [PubMed] [Google Scholar]
- 16.Vuong LT, Blood AB, Vogel JS, Anderson ME, Goldstein B. Applications of accelerator MS in pediatric drug evaluation. Bioanalysis. 2012;4(15):1871–1882. doi: 10.4155/bio.12.173. [DOI] [PubMed] [Google Scholar]
- 17.Lappin G, Shishikura Y, Jochemsen R, Weaver RJ, Gesson C, Brian Houston J, et al. Comparative pharmacokinetics between a microdose and therapeutic dose for clarithromycin, sumatriptan, propafenone, paracetamol (acetaminophen), and phenobarbital in human volunteers. Eur J Pharm Sci. 2011;43(3):141–150. doi: 10.1016/j.ejps.2011.04.009. [DOI] [PubMed] [Google Scholar]
- 18.Tozuka Z, Kusuhara H, Nozawa K, Hamabe Y, Ikushima I, Ikeda T, et al. Microdose study of 14C-acetaminophen with accelerator mass spectrometry to examine pharmacokinetics of parent drug and metabolites in healthy subjects. Clin Pharmacol Ther. 2010;88(6):824–830. doi: 10.1038/clpt.2010.206. [DOI] [PubMed] [Google Scholar]
- 19.Allegaert K, Tibboel D. Comments on “shift from biliary to urinary elimination of acetaminophen-glucuronide in acetaminophen-pretreated rats”. J Pharmacol Exp Ther. 2006;316(2):966–967. doi: 10.1124/jpet.105.096024. [DOI] [PubMed] [Google Scholar]
- 20.van der Marel CD, Anderson BJ, van Lingen RA, Holford NH, Pluim MA, Jansman FG, et al. Paracetamol and metabolite pharmacokinetics in infants. Eur J Clin Pharmacol. 2003;59(3):243–251. doi: 10.1007/s00228-003-0608-0. [DOI] [PubMed] [Google Scholar]
- 21.Zhou XJ, Garner RC, Nicholson S, Kissling CJ, Mayers D. Microdose pharmacokinetics of IDX899 and IDX989, candidate HIV-1 non-nucleoside reverse transcriptase inhibitors, following oral and intravenous administration in healthy male subjects. J Clin Pharmacol. 2009;49(12):1408–1416. doi: 10.1177/0091270009343698. [DOI] [PubMed] [Google Scholar]
- 22.van Duijn E, Sandman H, Grossouw D, Mocking JA, Coulier L, Vaes WH. Automated combustion accelerator mass spectrometry for the analysis of biomedical samples in the low attomole range. Anal Chem. 2014;86(15):7635–7641. doi: 10.1021/ac5015035. [DOI] [PubMed] [Google Scholar]
- 23.Klein MV, Vaes WHJ, Fabriek B, Sandman H, Mous DJW, Gottdang A. The 1 MV multi-element AMS system for biomedical applications at the Netherlands Organization for Applied Scientific Research (TNO) Nucl Instr Meth Phys Res B. 2013;294:14–17. doi: 10.1016/j.nimb.2012.06.024. [DOI] [Google Scholar]
- 24.Higton D, Young G, Timmerman P, Abbott R, Knutsson M, Svensson LD. European Bioanalysis Forum recommendation: scientific validation of quantification by accelerator mass spectrometry. Bioanalysis. 2012;4(22):2669–2679. doi: 10.4155/bio.12.242. [DOI] [PubMed] [Google Scholar]
- 25.Hopkins CS, Underhill S, Booker PD. Pharmacokinetics of paracetamol after cardiac surgery. Arch Dis Child. 1990;65(9):971–976. doi: 10.1136/adc.65.9.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anderson BJ, van Lingen RA, Hansen TG, Lin YC, Holford NH. Acetaminophen developmental pharmacokinetics in premature neonates and infants: a pooled population analysis. Anesthesiology. 2002;96(6):1336–1345. doi: 10.1097/00000542-200206000-00012. [DOI] [PubMed] [Google Scholar]
- 27.Walson PD, Halvorsen M, Edge J, Casavant MJ, Kelley MT. Pharmacokinetic comparison of acetaminophen elixir versus suppositories in vaccinated infants (aged 3 to 36 months): a single-dose, open-label, randomized, parallel-group design. Clin Ther. 2013;35(2):135–140. doi: 10.1016/j.clinthera.2012.12.016. [DOI] [PubMed] [Google Scholar]
- 28.Mooij MG, de Koning BA, Huijsman ML, de Wildt SN. Ontogeny of oral drug absorption processes in children. Expert Opin Drug Metab Toxicol. 2012;8(10):1293–1303. doi: 10.1517/17425255.2012.698261. [DOI] [PubMed] [Google Scholar]
- 29.Ratnapalan S, Bona N, Chandra K, Koren G. Physicians’ perceptions of teratogenic risk associated with radiography and CT during early pregnancy. AJR Am J Roentgenol. 2004;182(5):1107–1109. doi: 10.2214/ajr.182.5.1821107. [DOI] [PubMed] [Google Scholar]
- 30.de Wildt SN, Taguchi N, Koren G. Unintended pregnancy during radiotherapy for cancer. Nat Clin Pract Oncol. 2009;6(3):175–178. doi: 10.1038/ncponc1320. [DOI] [PubMed] [Google Scholar]
- 31.Maeda K, Ikeda Y, Fujita T, Yoshida K, Azuma Y, Haruyama Y, et al. Identification of the rate-determining process in the hepatic clearance of atorvastatin in a clinical cassette microdosing study. Clin Pharmacol Ther. 2011;90(4):575–581. doi: 10.1038/clpt.2011.142. [DOI] [PubMed] [Google Scholar]
- 32.Gorski JC, Jones DR, Haehner-Daniels BD, Hamman MA, O’Mara EM, Jr, Hall SD. The contribution of intestinal and hepatic CYP3A to the interaction between midazolam and clarithromycin. Clin Pharmacol Ther. 1998;64(2):133–143. doi: 10.1016/S0009-9236(98)90146-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.